of
Molecular Properties
A Web page by Kendal E. and Patrick M.
This website was made to show the optimized geometry calculations of 3 unique molecules, as well as to demonstrate the other atomic properties that can be calculated using quantum mechanics modeling software. This project started by finding the lowest energy geometry optimization of O2 , H2F2 , and 1,1-diethyl-2,2 cyanine. To do this the molecules were first constructed using Avogadro, which was then exported to wxMacMolPlt software which was used to construct an input file. The input file was then loaded into GamessQ which created an input file which was sent to GAMESSUS which solved a series of equations to find the lowest energy state of the molecule which was exported in the form of a .log file. This file was then loaded into Jmol to form a 3d model which could be manipulated to show not just the geometry of the molecule, but its atomic orbitals, vibrational modes, as well as an array of other calculated properties. Each molecule was first calculated to a relatively low energy configuration by using the 6-31G basis set. This was followed by CCD and CCT calculations respectively to find reasonably accurate estimates of the optimized molecular geometry.
For our diatomic molecule O2 , 2 different 6-31G basis sets had to be calculated to determine whether there was spin contamination caused by an open shell molecule. This was done by comparing UHF calculations to RHF. The UHF optimization had a lower energy output compared to the RHF optimization, so UHF was used for the remaining calculations.
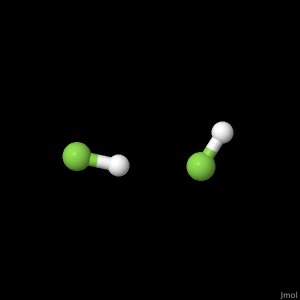
The polyatomic molecule we used was H2F2 which gave us the opportunity to observe how hydrogen bonding is calculated. This resulted in unique bond angles between the two HF portions of the molecule at the optimized geometric configuration, 168.7° and 121.0°. This closely echoed the results found in published scientific literature1. It was also noted that at lower energy calculations, the hydrogen bond length between the two HF portions of the molecule grew further apart.
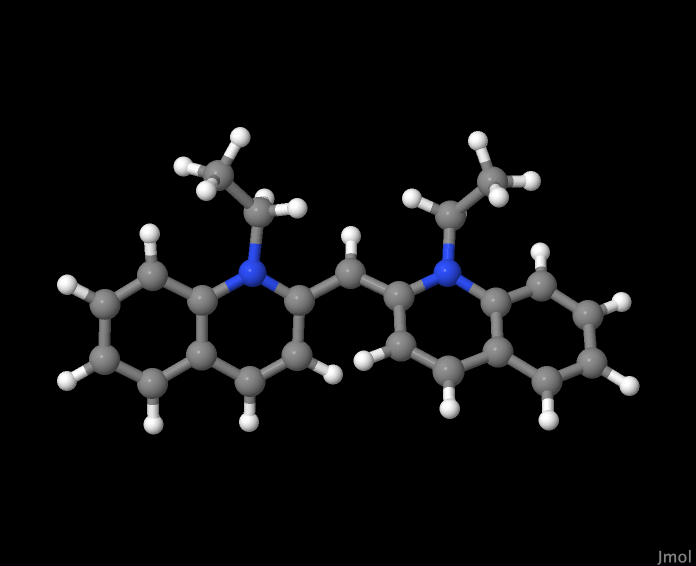
The Dye molecule 1,1-diethyl-2,2 cyanine had been used in a previous lab to calculate the bond length of the carbon chain by measuring the light absorption at different wavelengths, so it was naturally the first choice when looking for a large molecule to model. While we were unable to finish optimizing the molecule to its lowest energy geometry, we were able to gather a fair bit of information from the calculations that were performed.
 |
 |
 |
| Diatomic Molecule |
Poly-atomic Molecule - H2F2 |
Dye Molecule - 1,1-diethyl-2,2 cyanine |