Once the molecule file is fully loaded, the image at right will become live. At that time the "activate 3-D" icon ![]() will disappear.
will disappear.
Bimolecular Hydrogen Fluoride Compound
To create the diatomic hydrogen fluoride molecule, the structure
was built using the Avogadro software. A molecular mechanics geometry
optimization was ran to get it as close to its optimized configuration
before moving to a different software. MacMolPlt was then used to run a
RHF geometry optimization for a 6-31G basis set with its initial guess
set to Huckel. A CCD basis set was ran the same way using the 6-31G as a
starting point and finally a CCT basis set using the CCD as a starting
point. This concluded in the "best" optimized geometry for the molecule.
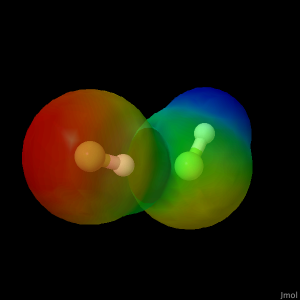
The
optimized geometry of the molecule was uploaded to Jmol where the
molecular electrostatic potential was mapped out onto the molecule's
surface.
Insert more information for “HF_ElectrostaticPotential” here.
The lengths of all of
the bonds were then calculated along with the angle of the three atoms
in the FHF bond that was being measured. The last hydrogen and the
fluoride it was attached to were translated together because the
measurement that was needed were for the other two bonds and their bond
angle. The second bond angle that is not displayed in the image that has
the green fluorine at its center has an angle of 121.0 degrees.
The four numbers that are labeled in green are the partial
atomic
charges for each atom. All of these calculations are for the molecule's
most optimized geometry. It is possible to manipulate the color of the
labels by right clicking on the structure it gives you when you choose
the bond length button and going to color>labels.
By pushing the button below
the highest occupied molecular orbitals for the compound are
highlighted. In the upper left corner of the picture shown for this
structure, there is the orbital that it is, 10 out of 20, and its
energy.
The HOMO is the last molecular orbital with the highest energy that contains electrons.
The button below allows you to see the the outlined orbitals of the
lowest unoccupied molecular orbital. Because the HOMO was the 10th
orbital, this one was the 11th. Its energy is also shown in the upper
left corner of the picture.
The LUMO is the orbital right above the HOMO in which it is the lowest orbital that does not contain any electrons.
There
were two separate vibrational frequencies calculated for the compound.
Each have their own animation although you can see that the two
different bonds of each hydrogen fluoride molecule more or less swap
motions. The vibrational frequency that corresponds to the animation
that appears when you press the button below was 4396.38cm-1.
The second vibrational frequency that correspond with the button below's animation was 4443.32cm-1.
The vibrational frequencies were calculated in MacMolPlt using
the molecule's optimized geometry. A numeric method for a Hessian run
was used for the vibrational analysis. Four other modes of vibrations
were also exhibited by this molecule. The two that are shown are
stretching vibrations which are the strongest and have the largest
frequency. The four others are all bending vibrations and the have
calculated frequencies such as 515.8cm-1, 441.64cm-1, 196.24cm-1, and
139.81cm-1.
You may look at any of these intermediate views again by clicking on the appropriate button.
Page skeleton and JavaScript generated by the Export to Web module of Jmol 14.31.24 2021-01-13 21:13 on Mar 1, 2021.