Baldomero, Miguel; Gilly, Shannon
Abstract
This study determines bond lengths, bond angles, dipole moments vibrational frequencies, and molecular orbitals using geometrically optimized molecule models of Flourine gas, Water, and Phenylacetylene. Different basis set sizes were used in optimizing geometries and were compared and contrasted. Results show that using the larger basis sets does not necessarily mean better results. Large basis sets results gave bond length, bond angles and vibrational frequencies that are close to the literature values. However, a smaller basis set is better in determining dipole moments.
Introduction
Determining the structure, geometry, and molecular orbitals of polyatomic molecules are vital entities in understand the behavior, reactivity, dipole moment, vibrational frequencies, polarizability, and other properties of molecules. Quantum mechanical principles can be used to describe molecular orbitals and structure of a molecule. Specifically, behavior of molecular orbitals can be condensed into wavefunctions. However, due to complexity of polyatomic molecules, approximations must be made to make an appropriate wavefunction. These approximations can be done by applying the variation principle which allows approximations of wavefunctions by using linear combinations of a trial wavefunction as shown in eq 1.
 (1)
(1)
This study determines bond lengths, bond angles, dipole moments vibrational frequencies, and molecular orbitals using geometrically optimized molecule models of Flourine gas, Water, and Phenylacetylene. Different basis set sizes were used in optimizing geometries and were compared and contrasted. Results show that using the larger basis sets does not necessarily mean better results. Large basis sets results gave bond length, bond angles and vibrational frequencies that are close to the literature values. However, a smaller basis set is better in determining dipole moments.
Introduction
Determining the structure, geometry, and molecular orbitals of polyatomic molecules are vital entities in understand the behavior, reactivity, dipole moment, vibrational frequencies, polarizability, and other properties of molecules. Quantum mechanical principles can be used to describe molecular orbitals and structure of a molecule. Specifically, behavior of molecular orbitals can be condensed into wavefunctions. However, due to complexity of polyatomic molecules, approximations must be made to make an appropriate wavefunction. These approximations can be done by applying the variation principle which allows approximations of wavefunctions by using linear combinations of a trial wavefunction as shown in eq 1.
The coefficients, c, are
then solved to produce the lowest energy for the electron.
Expectation value is then calculated and the wavefunction
is normalized. Each of the basis set wavefunctions used to
form the trial wavefunction is normalized. The larger the
basis set, the more accurate the energy prediction would
then be. In a geometry optimization calculation, one
searches for the best geometry to minimize the energy of
the system
In this study, geometry optimization are applied to fluorine gas, water, and phenyacetelyne molecule to determine bond-length, bond angles, dipole moment and many other properties. The main basis set used were 6-21G, 6-31G, and DZV, where DZV have the largest basis set, smallest for 6-21G.
Conclusion
Results show that using quantum mechanics to model structure and behavior of molecules is an effective way to determine physical quantities such as bond length, bond angles, dipole moments, vibrational frequencies, transition frequencies. However, certain basis sets are more effective in calculating a specific property or behavior. DZV, a large basis set, was effective in calculating bond lengths, bond angles, vibrational frequencies. Small basis sets, with diffused functions are more effective in determining dipole moments, electronegativity mapping, and partial charges.
References
Gutow, J. Molecular Orbital (MO) Calculations 2013, https://cms.gutow.uwosh.edu/Gutow/physical-chemistry-2/student-password-protected/lab-handouts/lab-handout-files/MO%20Calcs.pdf
Software:
-wxMacMolPlt
-GamessQ
-Jmol
-Seamonkey
In this study, geometry optimization are applied to fluorine gas, water, and phenyacetelyne molecule to determine bond-length, bond angles, dipole moment and many other properties. The main basis set used were 6-21G, 6-31G, and DZV, where DZV have the largest basis set, smallest for 6-21G.
| Fluorine
Gas |
Water |
Phenyacetylene |
 |
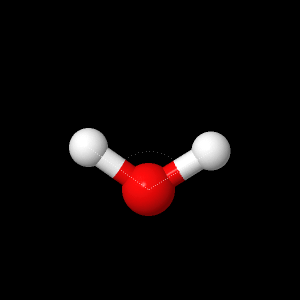 |
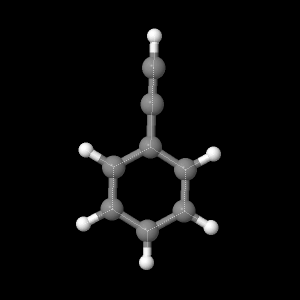 |
Conclusion
Results show that using quantum mechanics to model structure and behavior of molecules is an effective way to determine physical quantities such as bond length, bond angles, dipole moments, vibrational frequencies, transition frequencies. However, certain basis sets are more effective in calculating a specific property or behavior. DZV, a large basis set, was effective in calculating bond lengths, bond angles, vibrational frequencies. Small basis sets, with diffused functions are more effective in determining dipole moments, electronegativity mapping, and partial charges.
References
Gutow, J. Molecular Orbital (MO) Calculations 2013, https://cms.gutow.uwosh.edu/Gutow/physical-chemistry-2/student-password-protected/lab-handouts/lab-handout-files/MO%20Calcs.pdf
Software:
-wxMacMolPlt
-GamessQ
-Jmol
-Seamonkey