Quantum Calculations on Cl2, SCH2, p-Dichlorobenzene
Max Jakel, Chris Kempka
Abstract
In order to show the skepticism on the
regularity of data between different different levels of theory, the
properties of three molecules (Cl2, SCH3, 1,4-dicholorbenzen) were
calculated. Using Avogadro the three molecules were constructed and
had their geometries optimized in MacMol. From here, the vibrational
frequencies and HOMO orbitals were determined for Cl2 and SCH3 using
MacMol.
Introduction
Much of reaction chemistry depends on
the electronic structure of the molecules involved in a reaction.
The electronic structure of a molecule is its placement of electrons
in surrounding orbitals and the corresponding energies that relate
to them. Using this model of a molecule can also be used to predict
other properties of interest- such as the dipole movement,
vibrational frequencies, potential energy surface, and UV-Vis
spectrum calculations. These calculations, first done by hand by
physical and chemical physicists, can now be done faster and more
accurately with the use of computers and software packets. The use
of the computer analysis programs allow for companies to save a lot
of time and money by being able to determine if a reactions will
happen before it is tested. In addition, these programs generate 3-D
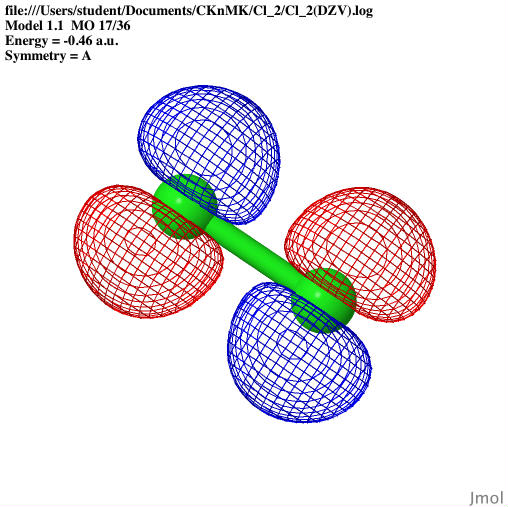
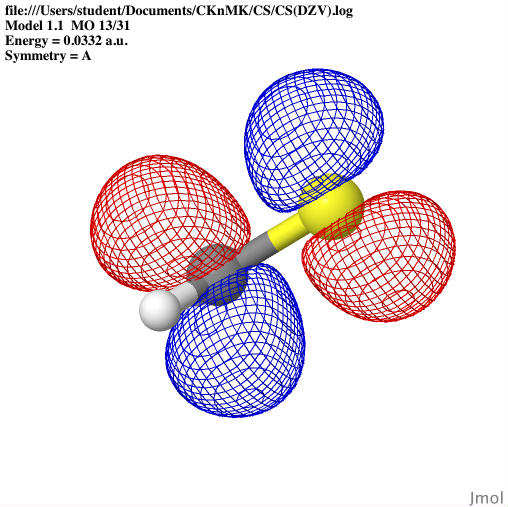
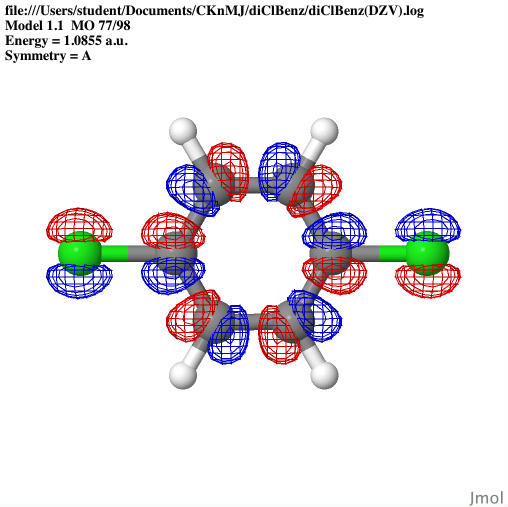
images that can show the: optimized geometries, HOMO orbitals,
vibrational frequencies, and dipole moment. However, one has to be
careful of what their calculations are reporting and how this
compares to what ab initio levels of theory were used. PM3
and ab initio levels of theories were used to compile a 3-D
image of dichloride, SCH3, and 1,4-dicholorbenzene and then the
optimized geometries were calculated using GamessQ. From these same
basis sets: the HOMO orbitals, dipole moment, and vibrational
frequencies were calculated for SCH3; the potential energy surface
and vibrational frequencies were calculated for Cl2; and a UV-Vis
spectra for the aromatic compound 1,4-dichlorobenzene.
Experimental
The program Avogadro
was used to create a digital image and get the initial geometry for
the three
molecules, Cl2, CH2S, and Dichlorobenzene.
Performing a molecular mechanics optimization in Avogadro gave the
initial
optimized geometry. After the geometry
was obtained, the file was moved to the program wxMacMolPlt in order
to prepare
the molecule for further optimization. Within
wxMacMolPlt each molecule was prepped to undergo calculations in
GamessQ by
setting up the correct basis sets for each level of theory.
The first of these basis sets was PM3, the
lowest level of theory, next was 6-21G where there were six Gaussian
functions
for the core electrons and 21 for the valence electrons, more on the
valence
because those are the electrons that participate in reactions.
6-31G, even more
Gaussian functions on the valence electron is a more accurate basis
set, and
lastly DZV or the Double Zeta Valence basis set was the most
accurate. These files were saved as .inp (input) files
to be opened in the program GamessQ, once the calculations were
completed
through GamessQ, they were checked to make sure optimal geometry was
reached. By viewing the log for each calculation and
checking to make sure that the calculations "exited gracefully" and
that the "Optimal Geometry Reached" was also present, the
calculation
completed correctly. After saving each
of these calculations as .log files, then opened in Jmol to view the
3D models.
The molecular orbitals were shown
through Jmol as well as the bond lengths and angles. The
dipole moments were taken from the GAMESSQ
files, opened in TextEdit, and each level of theory (6-21G, 6-31G,
and DZV) was
recorded. Vibrational frequencies for
Cl2 and CH2S were generated through wxMacMolPlt and calculated
through GamessQ. Potential Energy Surface vs Bond Length on
the diatomic molecule computations were prepped with the three
highest levels
of ab initio theory to be run in
GamessQ, then those files were placed into the Igor Tool supplied by
Dr. Jonathan
Gutow. For the aromatic,
Dichlorobenzene, UV-Vis spectra were completed through GamessQ.
Discussion
Looking at the data from each of the three molecules' quantum
calculations, it is evident that the calculations were best used for
calculating the (molecular) geometry and the resulting surface potential
energy plot. This is compared to vibrational frequencies which did not
match match very well with the experimental UV-Vis and experimental IR
values. These programs and methods have proven themselves over
time, so the sources of error would be first time use of said programs
and calculations.
References
1. COBLENTZ SOCIETY, IR Spectrum. 2009
http://webbook.nist.gov/cgi/cbook.cgi?ID=C106467&Units=SI&Type=IR-SPEC&Index=1#IR-SPEC,
March 9, 2014
2. NIST, Listing of experimental data for H2CS, (Thioformaldehyde)cccbdb.nist.gov/exp2.asp?casno=865361 March 9, 2014
3. J. Gutow, Molecular Orbital (MO) Calculations pp1-15. February 2014
4. J Gutow, Dr. Gutow's tools for Authoring Jmol Web Pages. June 11,
2010.
https://cms.gutow.uwosh.edu/gutow/Jmol_Web_Page_Maker/Jmol_Web_Page_Maker.shtml March 9, 2014