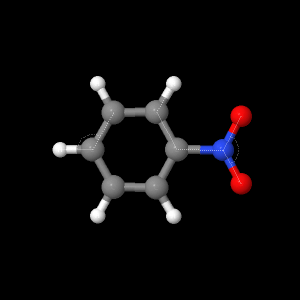
Nitrobenzene is an aromatic organic molecule with a
nitro-substituent group. The combustion
of the compound produces toxic nitrogen oxides, which can cause carcinogenic
effects when inhaled. As shown in the
figures to the right, the chemical structure and properties that classify this
molecule demonstrate its potential reactivity and ability to form other
compounds, such as aniline. The Lewis
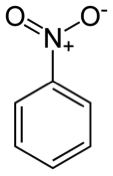
structure of the molecule is shown below.
To better understand the nature of the bonding and
anti-bonding molecular orbitals, three levels of theory were performed on an
initial guess of the molecular structure. These three levels (Molecular
Mechanics, MOPAC3, and Ab initio) served as the predication of the quantum
mechanical system for nitrobenzene.
By clicking on the button below, a figure of the molecule
can be viewed that shows the physical dimensions of the molecule.
The geometry of the molecule shows bond lengths between
carbon and hydrogen atoms as well as the bonding relationship with the
nitro-group. The partial negative charge
on one of the oxygen atoms and a partial positive charge on the nitrogen atom
depict the various molecular dipole moments calculated via the ab initio level
of theory. These dipole moments can be
correlated with UV-Vis transition energy as shown below in the table of dipole
moments from ground state to excited states.
Table 1. Cis-transition dipole moments of nitrobenzene from ground state to excited states. The calculations were performed using the optimized geometry of the molecule along with the double zeta valence (DZV) basis set. The unit of energy is Debye.
|
Excited state |
Transition energy
(Debye) |
|
1 |
0.012 635 |
|
2 |
0.124 501 |
|
3 |
1.227 428 |
|
4 |
2.899 312 |
|
5 |
2.283 397 |
|
6 |
5.232 152 |
|
7 |
4.333 443 |
|
8 |
0.241 692 |
|
9 |
3.578 363 |
|
10 |
4.580 314 |
The molecular dipole moments were initially calculated using an optimized geometry calculation and a variety of basis sets using ab initio level of theory. An experimental value, obtained from the NIST database, was found to be a value of 4.220 Debye. A table of the initial dipole moments prior to optimization is shown below.
Table 2. Initial dipole moments before optimization.
|
Basis set |
Dipole moment
(Debye) |
|
DZV |
5.853 971 |
|
6-21G |
5.229 080 |
|
6-31G |
5.813 726 |
|
AM1 |
5.147 224 |
|
PM3 |
5.248 982 |
Given the discrepancies between the experimental and
calculated values, the dipole moments were improved by adding a combination of
the following diffuse functions: D-heavy
atom polarization, F-heavy atom polarization, light atom polarization. The best optimization was determined to be a
combination of one D-heavy atom polarization and one light atom polarization
with a dipole moment of 5.087 767 Debye.
The electrostatic potential is shown as an all-encompassing
electric field surrounding the molecule. The color gradient of the field
represents the changes in the potential energy in relation to the charge
distribution. This attribute coincides with the partial atomic charges on
the nitro-substituent group.
The lowest unoccupied molecular orbital (LUMO) is shown
using the optimized geometry and DZV basis set. Molecular orbitals are shown
surrounding all atoms in the molecule, which is consistent with the molecular
orbital (MO) theory, where the LUMO is the lowest energy state of unoccupied
orbitals. According to the MO theory, delocalized electrons become
excited and transition from the HOMO to the LUMO. This transition energy
can be approximated using the ab initio level of theory.
By clicking on the button below, a figure of the molecule
can be viewed that shows a visual representation of the lowest unoccupied
molecular orbital (LUMO).
The optimized geometry and double zeta valence (DZV) basis
set were used to obtain the highest occupied molecular orbital (HOMO) for the
molecule. The balloon-like surfaces above and below each sulfur atom
represent electron occupied pi-orbitals.
By clicking on the button below, a figure of the molecule
can be viewed that shows a visual representation of the highest occupied
molecular orbital (HOMO).
The figure represents the stage when the valence electrons
are in the highest energy state. The red and blue-colored orbitals
represent the difference in phases for the pi and sigma orbitals.
Click here to view an animation of nitrobenzene.
Vibrational Motions
References:
1.
Mihalick, J. Gutow, J. Quantum Calculations 2016, p 1-12.
2.
Chlorine. Chlorine,
http://webbook.nist.gov/cgi/cbook.cgi?id=7782-50-5 (accessed Mar 3, 2016).
3.
Carbon disulfide. Carbon disulfide,
http://webbook.nist.gov/cgi/cbook.cgi?id=75-15-0 (accessed Mar 2, 2016).
4.
Benzene, nitro-. Benzene, nitro-,
http://webbook.nist.gov/cgi/cbook.cgi?id=c98953 (accessed Mar 3, 2016).