Quantum Calculation of Hydrogen Iodide, Formyl Fluoride, and p-Xylene
Andrew Balliet & Jaime Hernadnez
Abstract
Computational
models of hydrogen iodide (HI), formyl
fluoride (HFCO), and p-xylene (C8H10) were analyzed using
Hartree-Fock, self-consistent field computations (HF-SCF). The basis sets that were analyzed via the General
Atomic and Molecular Electronic Structure System (GAMESS) software were 3-21G,
SPK-DZP, and SPKr-TZP for hydrogen iodide, and 6-21G, 6-31G, and DZV for formyl
fluoride and p-xylene. The molecular
information of bond lengths and angles, vibrational frequencies, dipole
moments, etc. was calculated and compared to experimental values found from the
CRC Handbook of Chemistry and Physics
and the National Institute of Standards and Technology web book among
others. The basis sets that calculated
data closer to the experimental data were considered to be the best model used
for that particular molecule regardless of the size of the basis set. The most accurate model for hydrogen iodide
was SPK-DZP and DZV for formyl fluoride and p-xylene respectively.
Introduction
Mathematical models can very accurately calculate various characteristics of simple one-electron atoms. In that special case, the Hamiltonian term of the Schrödinger Equation for the system can be solved explicitly and quantifiable information such as the energy of the atom and the general location of the electron from the nucleus can be achieved. Equation 1 is the general form of the Schrödinger Equation, and Equation 2 is the Hamiltonian for a one-electron atom.¹
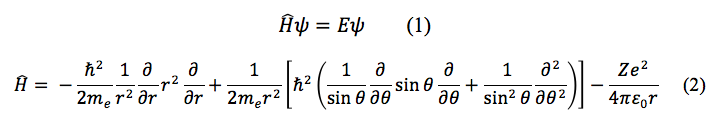
In the one-electron case, the
![]() term is completely separable, because there is no other
interactions between any other particles other than the electron and the
nucleus. When more electron are added to an atom, the electronic wavefunctions becomes impossible to solve since
the electron-electron repulsion term prevents the electrons from being
separable. To solve for this different
approximation to the many-electron atom Hamiltonian are used. The
electron configuration is one of the first approximations to the many-electron
wavefunction yet it doesn’t allow the distribution of one electron to change in
response to the position of another electron.
term is completely separable, because there is no other
interactions between any other particles other than the electron and the
nucleus. When more electron are added to an atom, the electronic wavefunctions becomes impossible to solve since
the electron-electron repulsion term prevents the electrons from being
separable. To solve for this different
approximation to the many-electron atom Hamiltonian are used. The
electron configuration is one of the first approximations to the many-electron
wavefunction yet it doesn’t allow the distribution of one electron to change in
response to the position of another electron.
To obtain calculated data
from more complicated atomic structures, the Hartree-Fock calculations are
used. These calculations are only
possible when the Born-Oppenheimer approximation is applied. The Born-Oppenheimer approximation states
that for an atom to possess electrons, the forces between the electrons and the
nucleus must be equal. Since the forces
between the electrons and nucleus are equal, the parametric qualities of the mass
and acceleration must combine to offset each other. Since the mass of a proton is at least three
orders of magnitude larger than the mass of an electron, the acceleration of
the electron must be equally as large to counter the nuclear mass to equalize
the force between the nucleus and the electron.
In a similar fashion to the application of the center of mass
relationship between the nucleus and the electrons the nucleus is so much more
massive that the mass of the atom is essentially the mass of the nucleus. Therefore since the electron has such a large
acceleration vector with respect to the nucleus in any new arrangement of the
nuclei, there would be an instantaneous electron response to the net electron
movement. With the Born-Oppenheimer
approximation the coordinates of the nucleus and electrons are considered
separable.²
The
many-electron wavefunctions calculations have more accurate results by the variational
method. Variational calculations adjusts the wavefunction in order to find the
minimum value for the energy of the ground state. So the lower the energy of
the adjustable wavefunction, the closer is to the correct answer. Most
variational methods are built initially on linear combination of atomic orbital
(LCAO) with several atomic orbital wavefunction where the coefficients are the
adjustable parameters in the wavefunction. In finding the expectation value for
the energy, the Hartree-Fock calculation are used to approximate the
electron-electron repulsion by averaging over all the positions of the other
electrons. ³
Another cornerstone to Hartree-Fock calculations is the application of Slater determinants and self-consistent field (SCF) approximations. A Slater determinant is the use by the determinate of a matrix to evaluate the one-electron wavefunctions of the ground state configuration to generate a many electron wavefunction of an atom. The SCF approximation is another application where the electron repulsion terms in the basis set are simplified to integrate, see Equation 3 for the general form of the multi-electron Hamiltonian. Where the summation term shows the sum of the electron repulsion wavefunction integrals.4
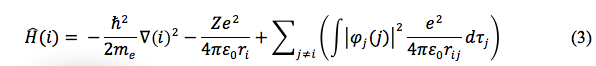
To
initiate the Hartree-Fock, self-consistent field computations (HF-SCF) for a
molecule, an initial geometry of the molecule must be used. Typically applying valence shell electron
pair repulsion (VSEPR) theory using a molecular modeling software package such
as AvogadroTM is a good initial arrangement. Then that geometric
data is transferred into a computational package such as The General Atomic and
Molecular Electronic Structure System (GAMESS) for HF-SCF computations.
Through
those complex and repetitive calculations, the wavefunctions and nuclei of the
molecule were adjusted to obtain the lowest electronic energy. When the lowest energy was achieved, that
particular geometry and other information such as vibrational frequencies, bond
length, etc. were reported. After that
information was obtained, higher basis sets were used to refine the molecular
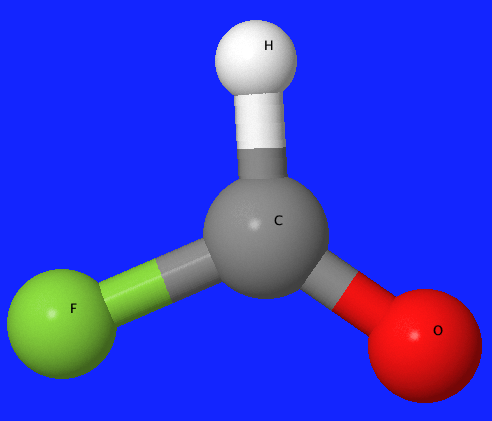
properties. In these particular calculations, hydrogen iodide (HI), formyl
fluoride (HFCO), and p-xylene (C8H10) were analyzed. The basis sets used for those molecules were
3-21G, SPK-DZP, and SPKr-TZP for hydrogen iodide, and 6-21G, 6-31G, and DZV for
formyl fluoride and p-xylene.
From these basis set and HF-SCF computations the calculated information obtained were the bond lengths and angles, electrostatic potential maps, dipole moments, partial atomic charges, and vibrational frequencies of the respective molecules. Additionally calculated information for the molecules includes the potential energy curve for the diatomic molecule and the UV-VIS absorption peaks for the aromatic compound. All of the calculated data and other information is displayed in the various pages on this website.
 |
 |
 |
| Hydrogen Iodide |
Formyl Flouride |
p-Xylene |
Experimental
The calculations were run on mac mini with intel i7 processor and 16G of RAM. The molecules
was first built using AvagadroTM.
After the molecule was built, the file was saved and opened into
wxMacMolPlt software to optimize the geometry of the molecule. In the case of hydrogen iodide, this was not
as important as hydrogen iodide is a linear diatomic molecule. However, in the subsequent models of formyl
fluoride and p-xylene, these geometry optimizations were much more
important. The purpose of the geometry
optimization of the molecule is for the use in the calculations that were
completed via GAMESS computation software to ensure the atoms in the
molecule were appropriately spaced, thereby reducing any unnecessary
calculation time. A MOPAC semi-emperical method was conducted by using either AM1 or PM3. After the geometry optimization of
the molecule was completed, the lowest level of theory was used
to calculate the molecular parameters for each one. Jmol was used in order to view the molecular orbitals, as well as geometry and vibrational information and visualization.
In the first level of theory
the Huckel approximation was used. The
Huckel approximation is an initial assessment that essentially only uses the
interactions between neighboring atoms in the Slater determinants that sets the
remaining interactions between non-neighboring atoms to zero. In addition, the overlap integral in the
determinate was also ignored. That left
only the diagonal echelon term of the matrix that was accounted for.
After the
lowest level of
theory was completed, in the case of hydrogen iodide this was 3-21G but
for formyl flouride and p-xylene this was 6-21G gaussian basis sets, The
results were then checked to ensure that the particle exited the
simulation
successfully and the experimental parameters were recorded. The
end of the Hartree-Fock, self-consistent
field (HF-SCF) method was then used for the next level of theory,
SPK-DZP for hydrogen iodide and 6-31G for formyl flouride and p-xylene,
to find a
better arrangement of the nuclei. After
the calculations were completed, the new optimized geometry was used in the
final level of theory. In hydrogen
iodide, that level was SPKr-TZP, in the other two molecules that was Double
Zeta Valence (DZV). The values that were
calculated were then compared to experimental parameters obtained from various
reference databases. The values that
were the closest to the reference databases were considered the best model
used.
Conclusion
In conclusion the Hartree-Fock, self-consistent field computations (HF-SCF) is a very useful and an efficient tool to calculate molecular parameters of the
three molecules when the computations were applied by the different approximations.
Hydrogen Iodide
The SPK-DZP basis set was
able to calculate the H-I bond length within 0.68% of the experimental
value. This basis set was not the
largest that was used to calculate the experimental parameters, but was the
closest overall in all of the calculated values compared to the experimental values. The SPKr-TZP was the largest overall basis
set used, with the 3-21G basis set being the smallest. One possible reason why this
basis set did not produce closer results to the experimental values was
specifically due to the atomic size of iodine. There were 53 electrons present
in the many levels of s, p, and d orbitals.
In that complexity, many of the basis sets that would have been used
such as 6-21G/6-31G and DZV basis sets were not available. However, with that complexity, HF-SCF
computations were almost required to accurately discern any molecular parameters
because of the amount of electrons present in the molecule. Even with this heteronuclear diatomic,
hydrogen iodide, the amount of electrons present had almost equivalent HOMO and
LUMO numerical orbitals in Jmol as the polyatomic aromatic p-xylene structure. In this particular case with the higher
amount of electrons present perhaps Density Functional Theory calculations
(DFT) would be slightly more accurate as DFT concentrates more on the electron
density between atoms. However, HF-SCF
was still very accurate even though the complexity of electron structure due to
the overall simplicity of the molecule.
With only two atoms, hydrogen and iodine, the dipole was relatively
straightforward.
One other important point to
note with the HF-SCF calculations was the HOMO and LUMO orbital displays. In the HOMO display, there were no
anti-bonding orbitals present unlike the p-xylene molecule. The HOMO figure illustrated the same general
bond character that the MO diagram estimated.
Formyl Fluoride
In the formyl fluoride
computations, the DZV basis set was the closest to the experimental
values. In all cases, this basis set was
within 1% of the experimental data obtained.
Due to this agreement between the computational and experimental data,
HF-SCF calculations were very useful and applicable to the molecular
structure. Similarly to hydrogen iodide,
formyl fluoride is a relatively straightforward polar molecule with a very established
dipole moment. Due to the high
electronegativity in the fluorine and oxygen ends, the density of electrons at
that end when compared to the more positive hydrogen side, lead to the very
linear dipole moment vector. Since this
was not a complicated molecule with conflicting electronegativities, the HF-SCF
showed a very good application of various parameters calculated.
As stated in the hydrogen
iodide molecule section, perhaps DFT would show a slightly better agreement
between the calculated and the experimental reference data, but with such a low
deviation, it would seem that the HF-SCF calculations were perfectly
applicable.
p-Xylene
In the p-xylene
simulation, this would almost require HF-SCF calculations at the very least to
determine any of the parameters.
In the
electrostatic potential map based on these calculations, the strongest regions
of electron density was inside the conjugated double bonds of the aromatic ring
which is qualitatively where it would be expected from any line angle diagram
of the molecule.
In the HOMO
display, there was such complexity in the aromatic region, that the
visualization of anti-bonding and bonding character was present. Specifically with this model, potentially DFT
would have been a more accurate basis set for calculation given the higher
electron density centered in the aromatic ring.
Refrences
1. Cooksey, A. Quantum Chemistry; Pearson: New York, 2014; Vol. 1, p 148.
2. Cooksey, A. Quantum Chemistry; Pearson: New York, 2014; Vol. 1, p 211-212.
3. Cooksey, A. Quantum Chemistry; Pearson: New York, 2014;
Vol. 1, p 194-195
4.Cooksey, A. Quantum Chemistry; Pearson: New York, 2014;Vol. 1, p 174-175.