Quantum Mechanics can be
used to figure out molecular properties which are essential to
knowing a molecules reactivity.These properties are; vibrational frequency, molecular
dipole moments, and polarizability.Using numerous
software packages such as Jmol, xmMacMolPlt, and Gamessq, all
of these properties can be calculated to a very high degree of
reliability which results in a very good understanding of how
certain molecules will react to other known molecules.
Introduction
A molecules electronic structure plays a
huge role in its reactivity.Knowing properties such as molecular dipole moments,
vibrational frequencies, and polarizability are key to knowing
a molecules reactivity.All
of these three properties are constantly being calculated by
chemical physicists and physical chemists to ensure the most
accurate literature values.With the help of computers loaded with software
packages, non-specialized individuals can do these
calculations with relative ease.Using this software geometry of structures can be
optimized and energy calculations can be computed to predict
whether reactions of certain compounds will work.Not only do these
programs generate useful data, they also generate 3-D models
of molecular structures, showing molecular orbitals as well as
vibrational movements for learning purposes.A key thing to
remember with these programs however is each level of theory
produces different results.The MOPAC uses empirical data as well as estimates the
values for two, electron overlap integrals.In MOPAC, PM3 and
AM1 are used as Hamiltonians.The best level of theory is ab initio.In ab initio all
integrals are calculated.All three theories, AM1, PM3, and ab initioare capable
of calculating optimized geometries, HOMO oribitals, dipole
moments, and vibrational frequencies.This web page will
show how the best levels of theory was chose based upon
calculated data for the molecular structures dinitrogen,
formaldehyde, and ethyl benzene.
Di-Nitrogen
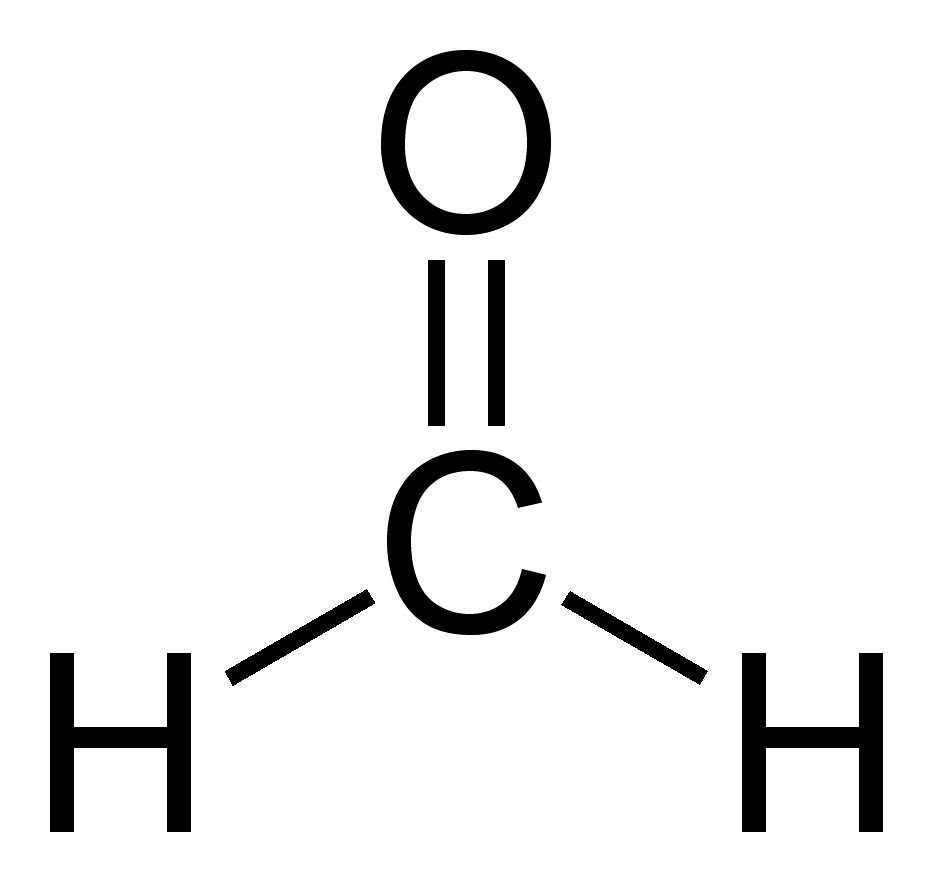
Formaldehyde
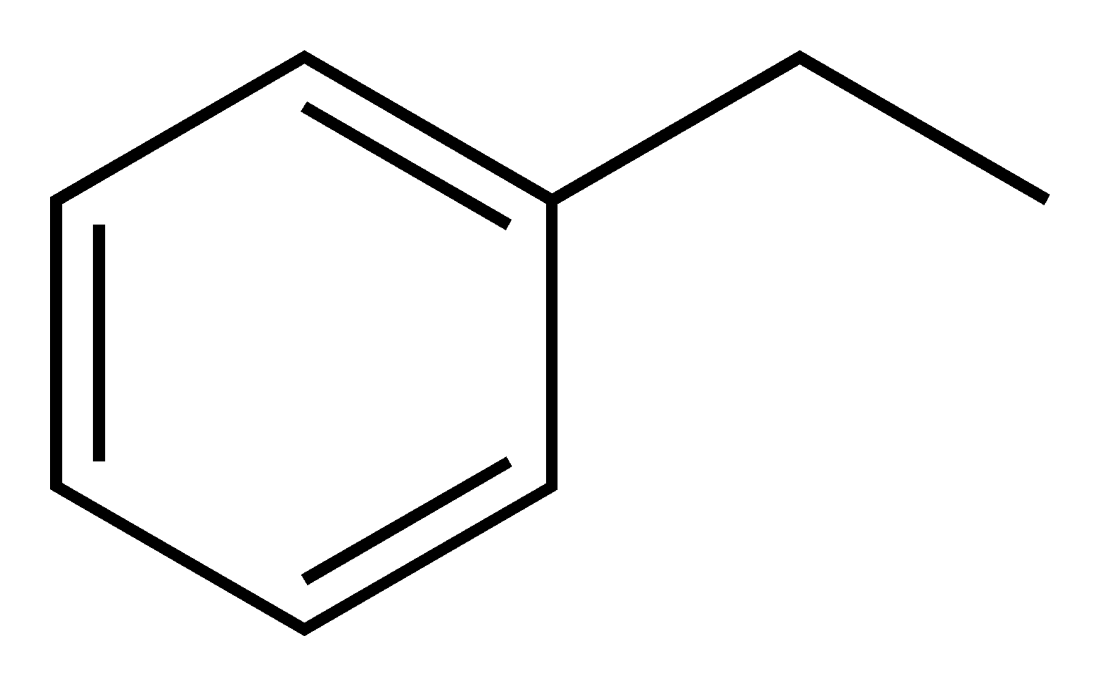
Ethyl Benzene
Conclusions
In conclusion, computational results have
great uses within the scientific world. These sorts of
calculations can be used to more closely study the properties
of molecules, especially new molecules, to understand and
predict how they will interact with other substances. However,
as much use these calculations have, there are weaknesses.
These sorts of calculations can be considered too high for
more simple systems and they may give heightened answers when
higher basis sets are used. For situations like that, other
calculations would be more appropriate. For example, taking in
geometries that include unpaired electrons and removing the
Huckel approximation will give better calculations as the
system becomes more realistic.