Molecular Orbital Calculations of
Bromobenzene, Hydrogen Cyanide and Nitrogen
By: Adam Dach and Logan OsbornIntroduction:
The molecular structure of molecules determines a great deal of its activity. When predicting reactivity of molecules, having the most optimized structure increases the prediction's accuracy. Characteristics such as vibrational frequencies, polarizability, dipole moment, electron donation and absorption probabilities can be predicted.The molecular orbitals in molecules can be described using wave functions. For a molecule with greater than one electron, solving for exact values is not possible. Operators can operate upon the wavefunction to extract useful information such as energy and position.
The three compounds being experimented upon are; Bromobenzene, Hydrogen Cyanide and Nitrogen1.
This experiment utilized the self-consistent field (SCF) theory for calculations. SCF is where an electron i is selected and its wavefunction Ψi
The optimization process was started with AM1 and PM3 because they were low level optimization, these were used to complete 321-G, which was used for 631-G, which was used for DZV optimization. This ordered successively improved the results while minimizing overall calculation time, since results from the previous level were used as a basis2.
Experimental:
The molecules are constructed using the program; wxMacMolPlt. The structures from this program were opened in Jmol and a mechanics optimization was performed. These files were saved as .xyz. WxMacMolPlt was again used to generate AM1 and PM3 geometry optimization input files (.inp) from the optimized .xyz files. GammesQ software was used to compute the results.The results were checked by opening the .log file and looking for a notification that the program "exited gracefully." This was an indicator that the calculations were completely successfully. These steps were repeated for Nitrogen, Hydrogen Cyanide and Bromobenzene. Bromobenzene calculations were started first because of the increased time they required. Jmol was used to see if the molecular orbitals calculations appeared logical compared with existing knowledge of pi and sigma bonds.
Geometry optimization was completed in order, starting with the AM1 and PM3 then going to 321-G, 631-G and finally Double Zeta Valence (DZV). wxMacMolPlt ws used to generate a vibrational frequency input file (.inp) which was computed using GamessQ. Dipole moments were extracted from the .log files by opening them in a text editing program and searching "debye" which were the units. The potential energy surface and bond length were plotted against each other for Hydrogen Cyanide, and optimized using 321-G, 631-G and DZV. The values were extracted using Jmol and a supplied macro.
Exterior Links:
Follow these links to view optimization and interactive molecular orbital diagrams
Results:
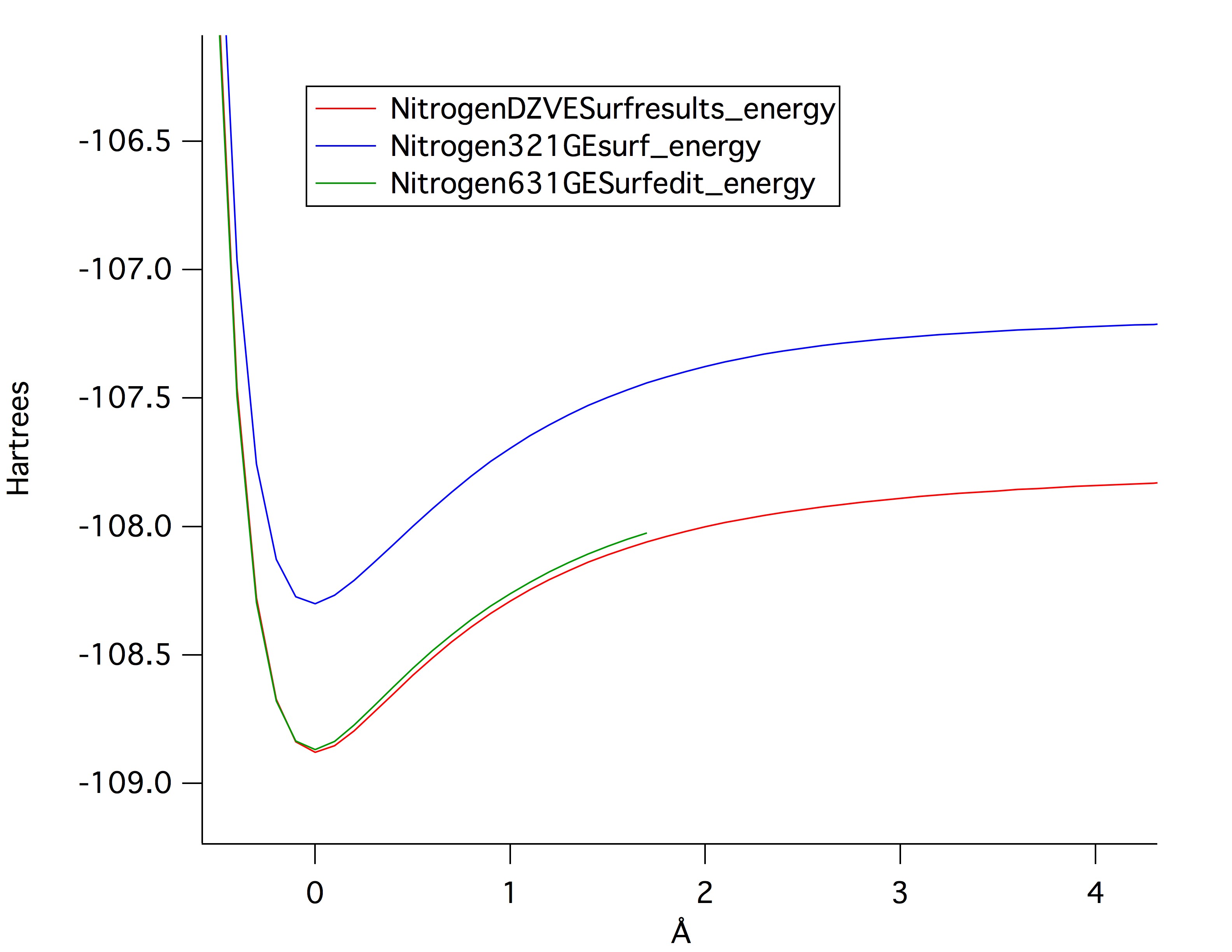
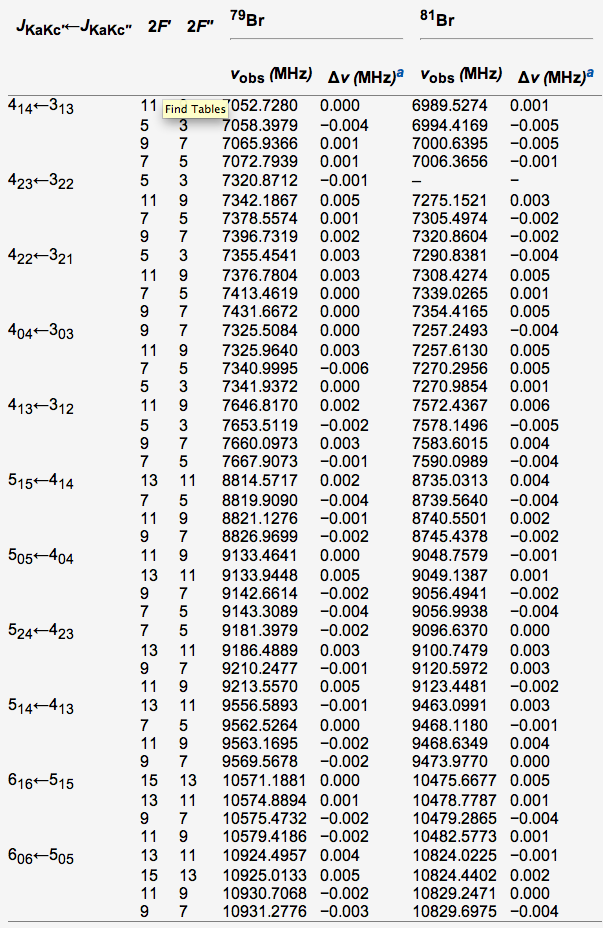
Figure 2. Table showing literature values for Bromobenzene3
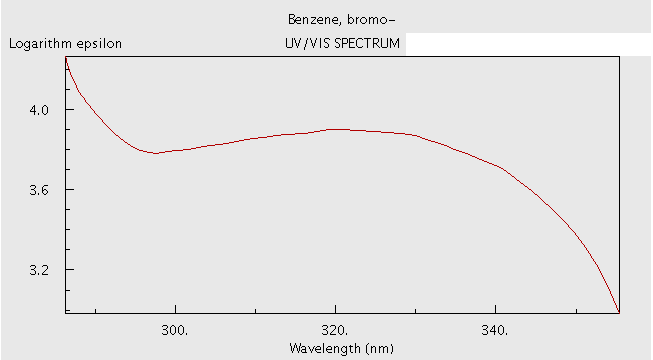
Figure 3. Graph of UV-VIS Spectrum for Bromobenzene4
Hydrogen Cyanide DZV Vibrational stretch frequencies from GamessQ optimization
Dx: -3.286949 Debye
Dy: .00604 Debye
Dz: 3.286954 Debye
Bromobenzene DZV Vibrational stretch frequencies from GamessQ optimization
Ground state: -.00073 debye
-2.169025 debye
-.000139 debye
This is slightly higher than the values recorded in this work of 119.4 degrees.
This same research shows bond lengths of 1.081angstrom.
This is again slightly higher than the 1.07 angstrom recorded in this work.
Conclusion: Computational results
are useful when data is difficult or impossible to obtain
experimentally and a prediction is difficult to produce by doing
calculations by hand. Additionally, computational results are cheaper
to generate; all one needs is a computer, a power socket, and time.
Computational calculations don't require a single reagent, so they are
also safer. On the other hand, spectroscopic data can sometimes be more
descriptive, accurate, or useful than computational results.
References
1. Gutow, Jonathan. "Chemistry 371 Lab Manual Spring 2011." (2011): 11-18. Print.
2. Atkins, Peter, and Julio De Paula. Physical Chemistry. 9thth ed. New York: W. H. Freeman and Company, 2010. Print.
3. Peebles, Sean, and Rebecca Peebles. "Determination of heavy atom structure of bromobenzene by rotational spectroscopy." Journal of Molecular Structure 657.1-3 Sept. (2003): 107-16. Web. 7 Mar. 2011.
4. Benzene, bromo-. National Institute of Standards and Technology, 2008. Web. 14 Feb. 2011.