Once the molecule file is fully
loaded, the image at right will become live. At that time the
"activate 3-D" icon ![]() will disappear.
will disappear.
F2 Quantum
Calculations
Electrostatic potential and partial atomic charges could not be predicted because this molecule consists of only fluorine atoms with a pure covalent bond. GamessQ and MacMolPlot measured a dipole moment of 0.00000 for all basis sets, which is to be expected.
Figure 1 shows a graph of potential energy vs. bond length for diatomic fluorine. As observed in the figure, the highest level of theory (DVZ), gives the lowest potential energy.

Figure 1: Potential Energy vs.
Bond Length for F2
Table 1 illustrates the molecular
orbitals present in fluorine.
Table 1: Molecular Orbitals
in Diatomic Fluorine
| Bonding |
Orbital |
| S sigma bonding |
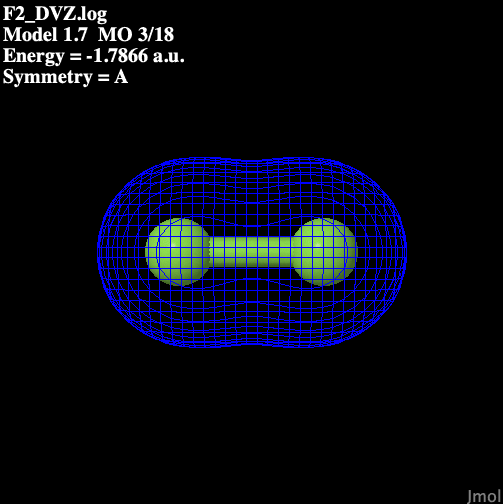 |
| S sigma anti-bonding |
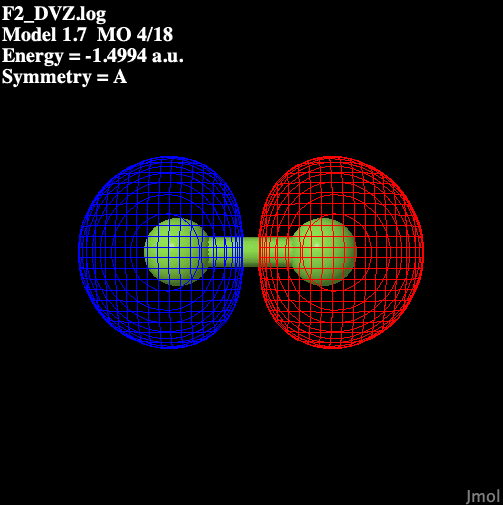 |
| P bonding |
 |
| P anti-bonding |
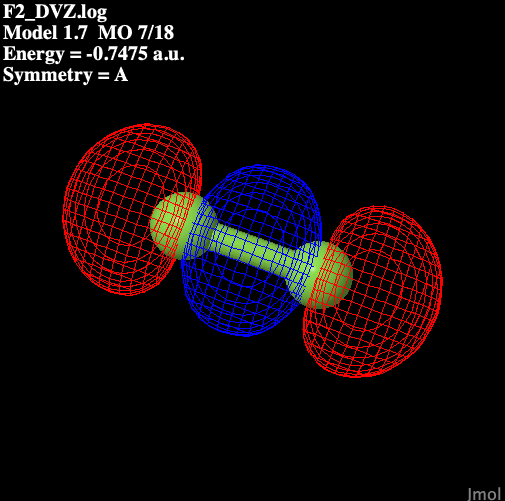 |
| P2 bonding |
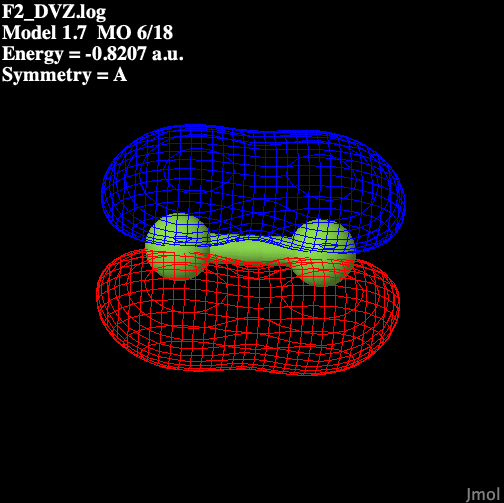 |
| P2 anti-bonding | 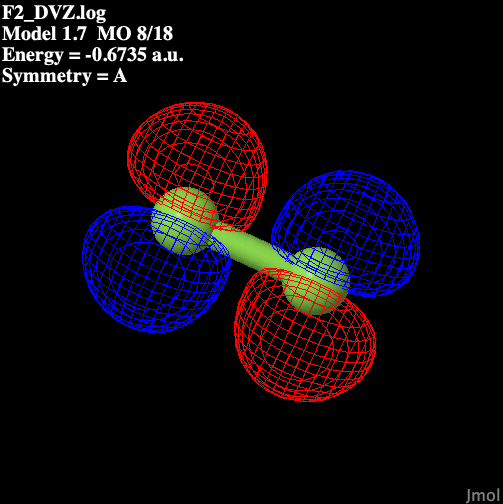 |
Page skeleton and
JavaScript generated by the Export to Web module of Jmol 14.29.46 2019-06-03
12:50 on Oct 3, 2019.
If your browser/OS combination is Java capable, you will get snappier performance if you use Java