Theoretical Calculations of Properties of Nitrobenzoic Acid
by Stacy Mundschau
Types of calculations performed
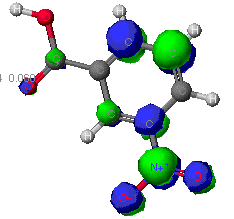
To generate the molecules you see below you, three separate calculations
involving the molecular oribitals and their associated electrons were performed
by CAChe software. Due to the complexity involved in describing these shapes
mathematically, assumptions concerning the extent to which these orbitals
interact had to be made. Each set of assumptions, refered to as the Extended
Huckel, MOPAC or ZINDO, varies in the treatment of these interacting, or
overlapping orbitals. While providing a simple method to generate molecular
values, the Extended Huckel theory can not match the agreement that MOPAC
and ZINDO calculations have with values determined in the laboratory. The
degree to which each theory accounts for the overlap between molecules
is a result of their dependence on a field of science known as quantum
mechanics. Particles decribed by quantum mechanics exist at the atomic
level (i.e electrons) and do not behave like the macroscale objects we
encounter. The superior accuracy of the ZINDO and MOPAC models is thus
a result of their reliance on quantum mechanical based modeling.
Results of geometry optimization at various levels of theory
The following depictions of nitrobenzoic acid are based on the types of
molecular orbital calculations performed.
While the figures above look very similar, the assumptions underlying
each of the associated calculations can result in dramatic differences
in the molecule's structure. The Extended Huckel theory treats molecules
as the familar balls linked to one another with sticks. The geometry the
molecule is governed by the bonds between balls acting as stiff springs.
ZINDO and MOPAC use the principles of quantum mechanics to determine the
most likely shape of the model, based on the lowest calculated energy value.
This results in a much more complete mathematical treatment of the molecule,
and thus provides more accurate molecular property values and depictions.
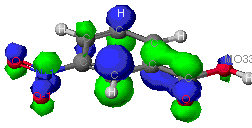
The molecular orbitals that provide the primary bonding
Not all of the molecular orbitals calculated by each model contribute equally
to the overall properties of nitrobenzoic acid. Each theory's calculations
essentially "weight" the significance of each orbital. The orbitals selected
below are by no means all of the molecular orbitals that exist. However,
their low energy (listed by the molecular orbital number) provides a more
stable environment for electrons to exist and interact with neighboring
nuclei and electrons. Selected orbitals from the ZINDO model that contribute
to the bonding in the aromatic ring are provided below.
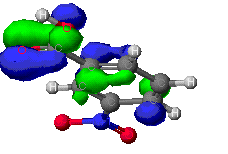
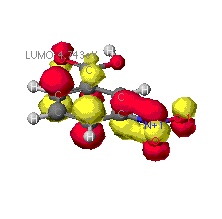
The highest occupied (HOMO) and lowest unoccupied (LUMO) molecular orbitals
The identification of the HOMO and LUMO molecular orbitals are critical
in understanding the manner in which a reaction takes place. As these are
the sites which contain the most energetically unstable electrons (HOMO)
and most willing to accept electrons (LUMO), it follows that these are
the orbitals most involved in chemical reactions . A common example of
a chemical reaction which can be used to illustrate the importance of HOMO
and LUMO orbitals is redox reaction. Consider the process of rusting in
which neutral iron atom loses electrons to oxygen. More specifically, the
redistrubution of electrons from the HOMO orbital of iron atom to the LUMO
orbtial of oxygen weakens the bonds between oxygen atoms. The decreased
bond strength between oxygen atoms drives the cleavage of the oxygen molecule
and the simultaneous oxidation of iron. In addition to explaining reactions,
the ability to identify HOMO and LUMO orbitals can be used to predict if
a proposed reaction will occur.
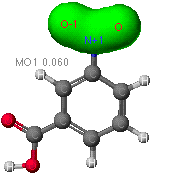
Electron density/distribution maps
In addition to determining the shape and energies of the molecular orbitals,
each theory is able model how the electrons will arrange themselves over
the molecule. That is, these calculations will show where the electrons
will most likely be in a concerted effort to lower the overall energy of
the molecule. resonant to alternating positions in the molecule.
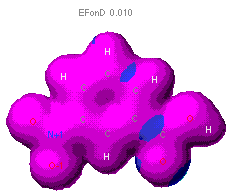 |
The diagram of nitrobenzoic acids how electron density
is centered within the aromatic ring. (Recall that aromatic rings are molecules
commonly represented by rings whose double bonds "flip" or resonant to
alternating positions in the molecule.) As electrons carry a negative charge,
the increased probability that they will be found in a certain region of
the molecule effectively polarizes the molecule. The blue reg |
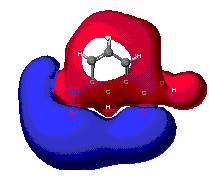
Charge distribution maps
This tendency of electrons to polarize certain regions of the molecules
allows for the intramolecular (inside of the molecule) charge to be depicted.
It is important to mention that the prescece of negatively charge regions
requires that equally postive charged regions must exist if the overall
molecule is going to remain neutral.
So why does electron redistribution to certain regions of the molecule
result in lower energies than in other arrangements? Essentially what accounts
for this hoarding of electrons into concentrated regions is a property
of individual atoms known as electronegativity. The tendency of electrons
to gather near highly electronegative atoms is related to the strength
of attraction to its nucleus. Small molecules with with few electrons "shielding"
other electrons from its nucleus give rise to the most electronegative
atoms. This pattern is reflected in the general increase of electronegativity
moving up the periodic table and to the right (excluding the noble gases).
Notice in the diagrams below that the blue colored regions correspond to
the highly electronegative oxygen molecules. Their eletronegativity decreases
the charge of the neighboring molecules. The unequal sharing of electrons
between molecules results in partial atomic charges, or the charge that
one atom experiences. The polarity of nitrobenzoic acid is a direct result
of partial atomic charges in the region.
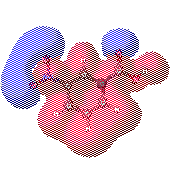
|
|
Calculated dipole moments
By determing the molecular orbitals and regions of chrage distribution
through mathematical models, calculating overall the dipole moment of the
molecule is possible. The dipole moment is a vector that shows how the
charge is distributed in a molecule. It can be thought of as the net sum
of the individual dipoles within the molecule. The following table lists
the dipole moment values calculated by the type of model structure.
| Extended Huckel |
8.967 |
| ZINDO |
5.417 |
| MOPAC |
5.633 |