Theoretical Calculations of Properties of Benzoic Acid
by Dustyn Sawall
Types of calculations performed
In general, Molecular orbitals (MO) are combinations of Atomic
orbitals (AO). Atomic orbitals are actually mathematical expressions
representing where a electron in a given quantum state can be found.
When AO's are added together, they are done so in a way to minimize
the energy of the system. This is done by some contributions of AO's
being greater than others. The mathematical formula for this
simplifies into three type of factors, the energy of the original AO,
the energy saved by forming the bond and an overlap factor dependent
on multiple interactions between electrons that need to be evaluated
at the same time.
These equations rapidly become huge. To do them for all of the
overlap considerations takes considerable time and computing power.
Even then, they don't all work out. These are known as ab initio
calculations. To improve workability of the mathematics, some
modifications can be made. Generally, the more of the overlap
integrals you use, the better your calc, but that is a trade off with
if they can be done at all.
In the programs used for this page, three different levels of
theory are analyzed. In the extended Huckel theory, it is assumed
that only nearest neighbors interact. Zindo level of theory uses
intermediate neglect of the overlap intergrals. It works using some
empirical data from ab initio calculations to optimize its MO's.
MOPAC level of theory uses a moderate neglect of these intergrals and
uses empirical data from thermodynamics to improve it's theory. MOPAC
is the most precise of the programs used because it takes into
account more of the intergrals in question.
Results of geometry optimization at various levels of theory
Initially, only a Molecular Mechanics program was used. This
program treats the atoms as classical harmonic oscillator. The bond
lengths and angles are calculated from rotational and vibrational
spectroscopy results. The computer has tables of bond lengths, the
computer then uses standard values from these tables to optimize the
molecules. The molecule is designed so as to minimize the stress
between atoms. This is the program used for the extended huckel level
of theory. Zindo and MOPAC have geometry optimizations available to
them from quantum theory to further optimize the molecules. In
theory, the Mopac calc should be best. If the bond lengths and
angles were examined in detail, there would be slight differences,
but as can be seen from the pictures below, they are slight.
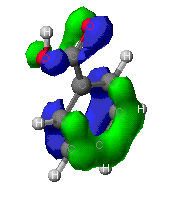
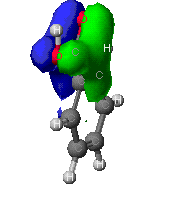
The molecular orbitals that provide the primary bonding
Here are a few of the molecular orbitals for toluene. This is only
two of the orbital that provide bonding fro the molecule, there are
several others. As can be seen, the MO's can cover large portions of
the molecule.
|
This is an example of a sigma bond from the carbon atoms
throughout the benzene ring.
|
This is one veiw of the pi bond formed from the p orbitals
in the carbons of the benzene ring.
|
This is simply another veiw of a pi bond. Notice how the
orbital extends in to the co-planar areas of the acid
group.
|
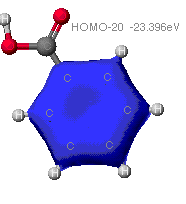
The highest occupied (HOMO) and lowest unoccupied (LUMO)
molecular orbitals
These are the orbitals that are most involved in chemical
reactions since they can most easily lose or gain electrons.
|
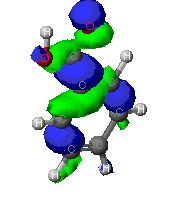
Extended Huckel
|
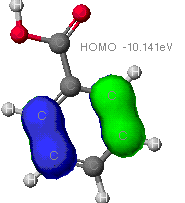
MOPAC
|
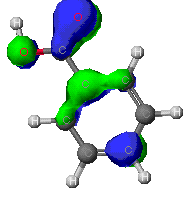
ZINDO
|
|
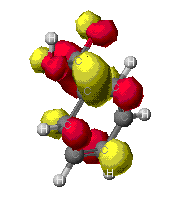
|
|
|
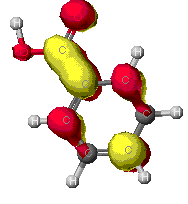
As can be seen from these diagrams, the extended
Huckel theory and the other two often differ. This is because of the
different assumptions each make. In general the MOPAC level of theory
is superior. These show the orbitals that are at the Highest Occupied
MO, that is the MO that is occupied at an elevated energy level. The
red/yellow is the Lowest Unoccupied MO. There are no electrons in
these MO's as they are at such an elevated energy level.
Electron density/distribution maps
|
 Extended Huckel Extended Huckel
|
 MOPAC MOPAC
|
 Zindo Zindo
|
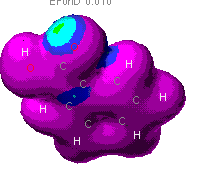
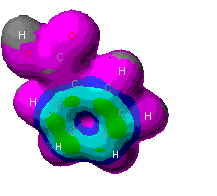
This is diagram showing the probability of finding an
electron in the given areas of map, the warmer the color, the more
likely that an electron could be found there. As can be seen,
there is a great discrepency between the levels of theory. It is
expected to find electron density in the pi orbitals of the ring, as
in the MOPAC theory. It would also be expected to find the highest
concentration around the oxygens in the models though. This is seen
in the Zindo image. For this reason, the ZINDO model looks most
probable.
Charge distribution maps
|
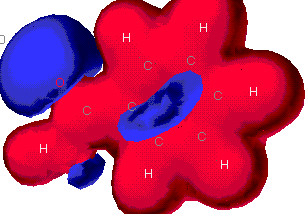 MOPAC MOPAC
|
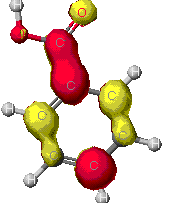
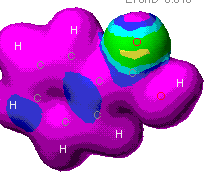
These maps show the relative distribution of positive
and negative charge on the molecules. The red indicates a positive
characteristic and blue is for a negative characteristic. This
diagram shows mostly a positive charge around the benzene ring, but
there is a strong negative charge near the acidic end. A
partial negative charge will be found on the acid end of the molecule
and a partial positive charge will be distributed around the ring.
Calculated dipole moments
|
Chemical Sample
|
Dipole Moment
|
|
Benzoic Acid via Extended Huckel
|
4.086
|
|
Benzoic Acid via Mopac
|
2.254
|
|
Benzoic Acid via Zindo
|
1.717
|
Sep_Lab_Home.html to
return to the main page