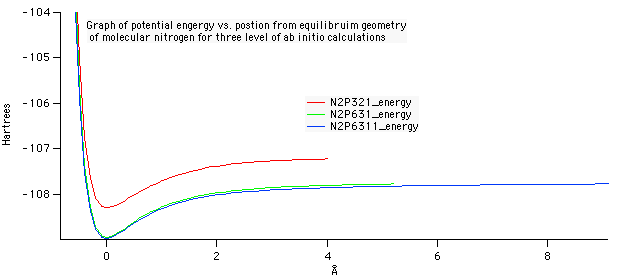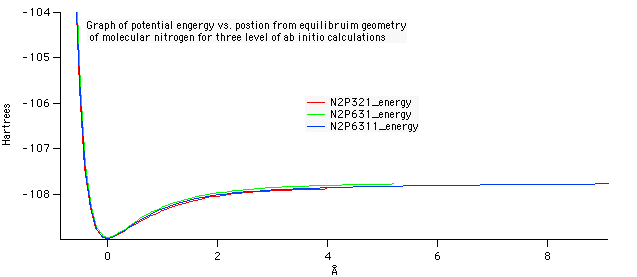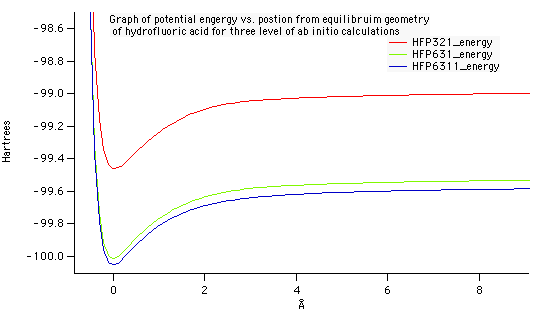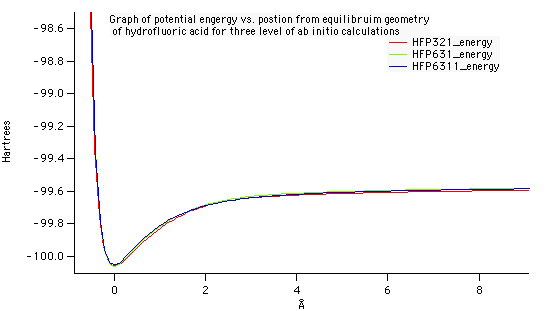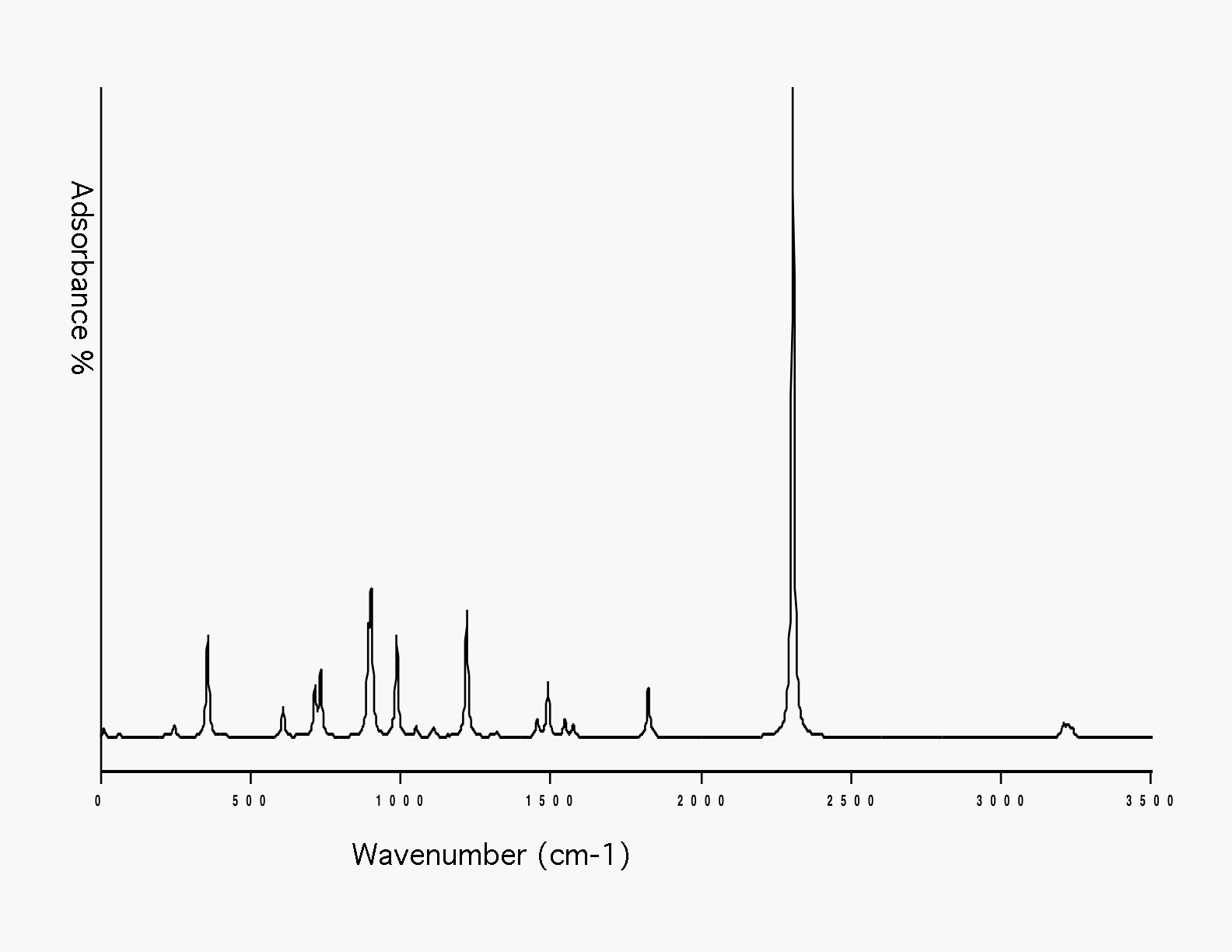
Optimized geometry for nitrogen at the 6-31G level of theory. A bond length of 1.083 Angstroms was compared against the experimental data on the NIST website. The accuracy of this value to the experimental was what determined 6-31G to be the best level of theory.