By Brooke Koshel and Cassidy Sprenger
Introduction:
Molecular orbitals are wavefunctions for electrons in molecules. Eigenfunctions and eigenvalues are only found when a single atom with one electron is used. For potential energy, the Hamiltonian operator becomes more complicated for systems with more than two charged particles, and no analytical solutions have been found for a eigenvalue. The following variational principle shows how to approximate true wavefunctions with sums of trial wavefunctions.
Calculations were carried out on fluorine, carbon monoxide, and benzoic acid. Different software is available for doing these calculations. Ab initio theory software calculates all integrals and is the best level of theory. Different basis sets of increasing size are available for use. Vibrational frequencies were determined for each molecule, UV-Vis was done for benzoic acid, and potential energy was determined for both diatomic molecules.
Fluorine:
The RHF6-31G(d,p) level of theory was selected as the best level of theory for optimization based on the comparison of experimental bond length to those calculated from the NIST website. Figure 1 shows optimized geometry.
Molecular orbitals are wavefunctions for electrons in molecules. Eigenfunctions and eigenvalues are only found when a single atom with one electron is used. For potential energy, the Hamiltonian operator becomes more complicated for systems with more than two charged particles, and no analytical solutions have been found for a eigenvalue. The following variational principle shows how to approximate true wavefunctions with sums of trial wavefunctions.
Ψ = c2 Ψ2 + c2 Ψ 2 + c3 Ψ 3 + ...
(Equation 1)
In equation one Ψ I is a trial wavefunction and ci
is its contribution to Ψ. To find the set of coefficients which
produces the lowest energy for the electron, the expectation value of
the energy is calculated by using the following equation:<H> = <E> = ΙΨ*H Ψdτ/Ψ*Ψdτ
= c2 ΙΨ*2H Ψ2 dτ +c2c2ΙΨ*2H Ψ2 dτ + c2c3ΙΨ*2H Ψ3 dτ +.../ c2 ΙΨ*2 Ψ2 dτ
+c2c2ΙΨ*2Ψ2 dτ + c2c3ΙΨ*2 Ψ3 dτ +...
The total wavefunction is not normalized, which in
turns puts more constraints on the values of the coefficients. To
get a more accurate energy prediction, the basis set has to be
larger. An accurate molecular geometry is needed for a correct
prediction of electronic energy. “A geometry optimization
calculation is defined as one searches for the best geometry by trying
different arrangements of atoms until the energy of the system is
minimized” (1). Calculations were carried out on fluorine, carbon monoxide, and benzoic acid. Different software is available for doing these calculations. Ab initio theory software calculates all integrals and is the best level of theory. Different basis sets of increasing size are available for use. Vibrational frequencies were determined for each molecule, UV-Vis was done for benzoic acid, and potential energy was determined for both diatomic molecules.
Fluorine:
The RHF6-31G(d,p) level of theory was selected as the best level of theory for optimization based on the comparison of experimental bond length to those calculated from the NIST website. Figure 1 shows optimized geometry.
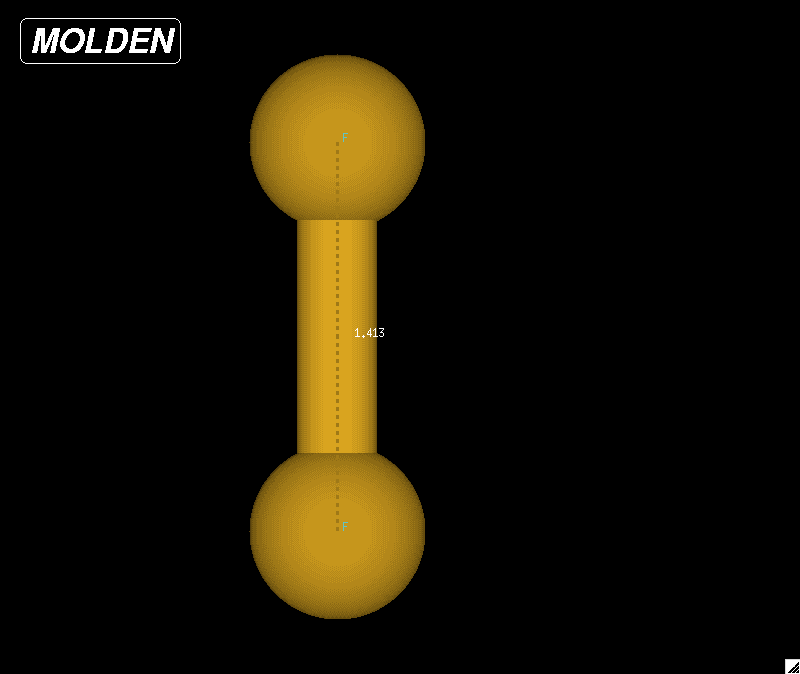 |
| Figure 1: Optimized geometry for a diatomic fluorine molecule. The bond length was determined to be 1.41252 Angstroms based on these calculations. The NIST website reported a bond length of 1.412 Angstroms. |
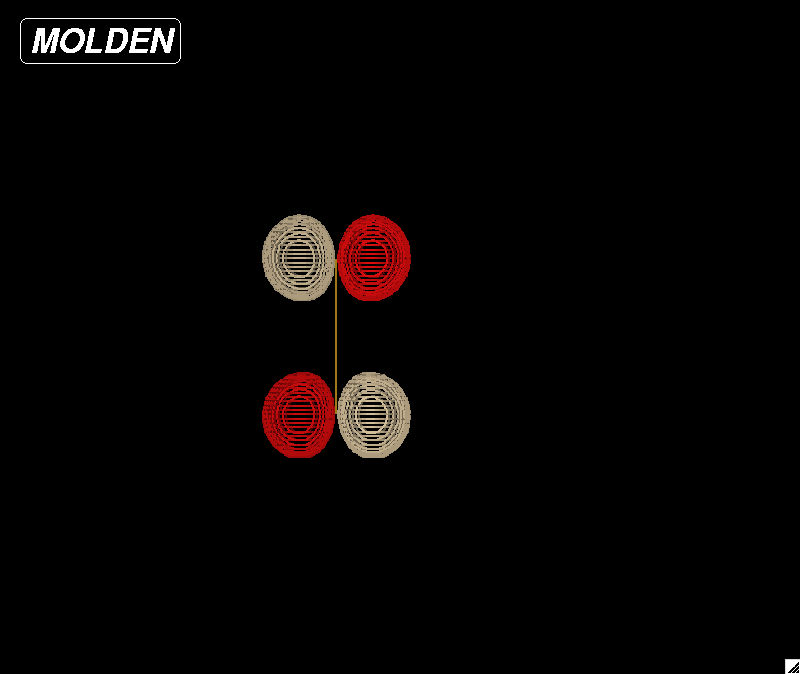 |
| Figure 2: HOMO (highest occupied
molecular orbital) for a fluorine molecule. |
According to the
RHF6-31G(d,p) level of theory, the vibrational frequency was determined
to be 1013.27 1/cm for the stretching vibration between the two
atoms. The NIST website reported a frequency of 917 1/cm.
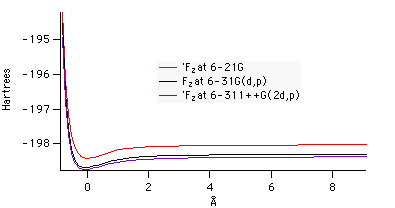 |
| Figure 3: Potential energy
combined plot at various levels of theory. |
Figure 3 shows that
as the basis set increases, potential energy decreases.
Carbon Monoxide:
The RHF6-21G(d,p) level of theory was selected as the best level of theory for optimization based on the comparison of experimental bond length to those calculated from the NIST website. Figure 4 shows optimized geometry.
According to the RHF6-21G level of theory, the vibrational frequency was determined to be 2295.80 1/cm for the stretching vibration between the two atoms. The NIST website reported a frequency of 2170 1/cm.
Figure 6 shows that as the basis set increases, potential energy decreases. By comparing the plots of potential energy for fluorine and carbon monoxide, one can see that the energy is higher for carbon monoxide at each corresponding level of theory. As energy increases, stability decreases, so these results imply that fluorine is a more stable diatomic than carbon monoxide.
Benzoic Acid:
The RHF6-311G(d,p) level of theory was selected as the best level of theory for optimization based on the comparison of experimental bond length to those calculated from the NIST website. Figure 7 shows optimized geometry.
According to the RHF6-311G(d,p) level of theory, the vibrational frequency are shown in the following table.
Table 1: Shows the calculated vibrational frequencies for different
atom bonding for benzoic acid and compares the calculated values to
values shown on the IR spectrum from the NIST website, the IR
spectrum can be found at the following link: http://webbook.nist.gov/cgi/cbook.cgi?ID=C65850&Units=SI&Type=IR-SPEC&Index=0#IR-SPEC
Table 2: Shows the UV-Vis calculations using the 6-21 and 6-31 levels of theory. The data was compared to a UV-Vis spectrum from the NIST website. It was noticed that the calculated data fit into the same relative shape of the spectrum, however, the calculated excited energy states were too high. To see the NIST spectrum log on to: http://webbook.nist.gov/cgi/cbook.cgi?ID=C65850&Units=SI&Mask=400#UV-Vis-Spec
Conclusion:
The programs used in this experiment were useful in determining information that would be very difficult to calculate by hand. Computers also have the luxury of being able to use large basis sets and perform calculations on complex molecules. Unfortunately, data reported does not always provide very accurate results, which can be seen in our calculations above. Computer simulations are useful for student laboratory, but not reliable for industrial laboratory type settings.
References:
Gutow. "Molecular Orbitals Experiment". 9/05. Pg. 1
NIST website
Lide, D.R. The CRC Handbook of Chemistry and Physics. 81ed. New York: CRC Press, 2000.
Brooke_Cassidy/371_Protected/M01_Lab
Carbon Monoxide:
The RHF6-21G(d,p) level of theory was selected as the best level of theory for optimization based on the comparison of experimental bond length to those calculated from the NIST website. Figure 4 shows optimized geometry.
 |
| Figure 4: Optimized geometry for a carbon monoxide molecule. The bond length was determined to be 1.13059 Angstroms based on these calculations. The NIST website reported a bond length of 1.1283 Angstroms |
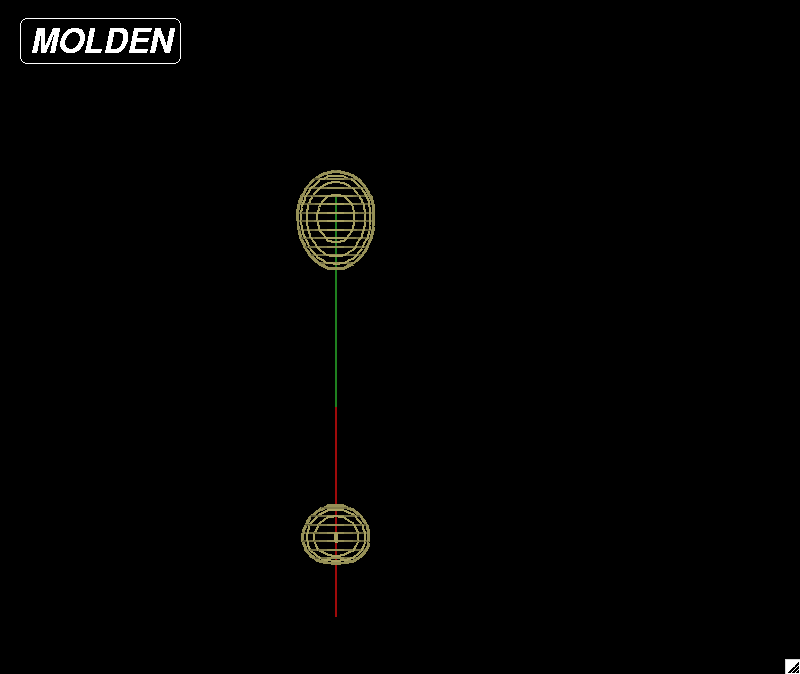 |
| Figure 5: HOMO (highest occupied molecular orbital) for a carbon monoxide molecule. |
According to the RHF6-21G level of theory, the vibrational frequency was determined to be 2295.80 1/cm for the stretching vibration between the two atoms. The NIST website reported a frequency of 2170 1/cm.
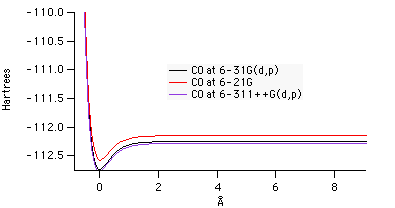 |
| Figure 6: Potential energy combined plot at various levels of theory. |
Figure 6 shows that as the basis set increases, potential energy decreases. By comparing the plots of potential energy for fluorine and carbon monoxide, one can see that the energy is higher for carbon monoxide at each corresponding level of theory. As energy increases, stability decreases, so these results imply that fluorine is a more stable diatomic than carbon monoxide.
Benzoic Acid:
The RHF6-311G(d,p) level of theory was selected as the best level of theory for optimization based on the comparison of experimental bond length to those calculated from the NIST website. Figure 7 shows optimized geometry.
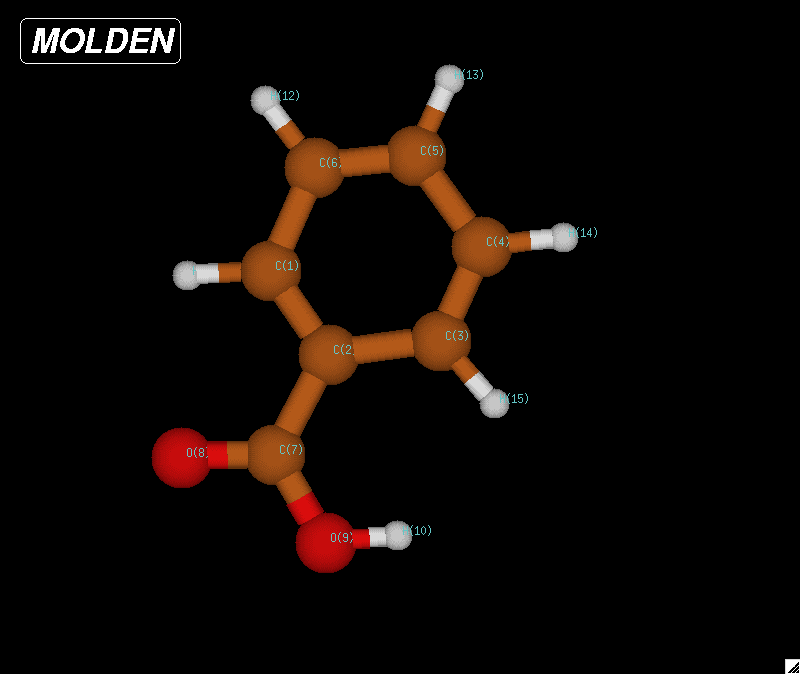 |
| Figure 7: Optimized geometry for
a benzoic acid molecule. The
bond lengths were determined to be 1.08977 Angstroms for C-H, 1.33631
Angstroms for C=C based on these
calculations. The CRC Handbook reported a bond length of 1.084
Angstroms for C-H, 1.395 Angstroms for C=C. |
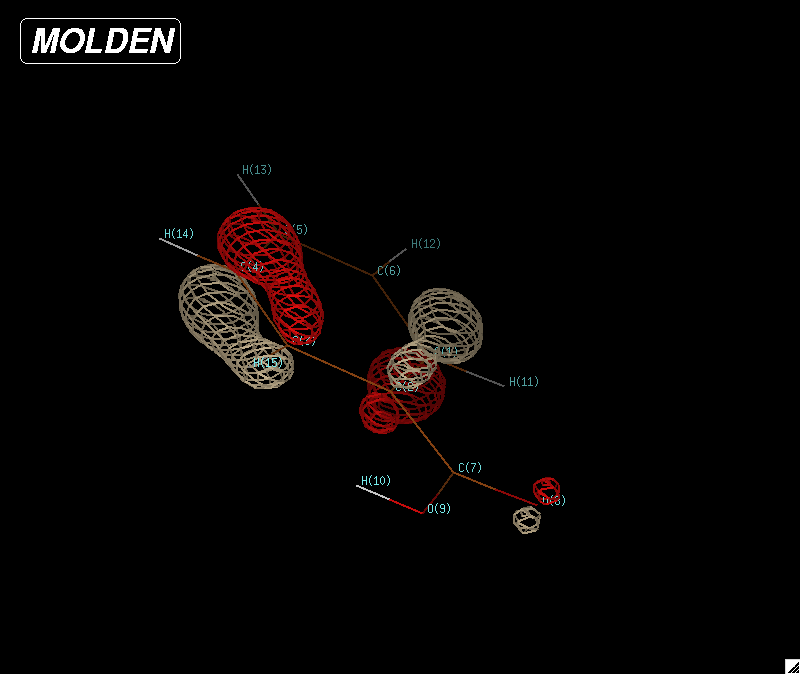 |
| Figure 8: HOMO (highest occupied molecular orbital) for a benzoic acid molecule. |
According to the RHF6-311G(d,p) level of theory, the vibrational frequency are shown in the following table.
| Calculated
Vibrational Frequencies (1/cm) |
NIST Vibrational Frequencies
(1/cm) (From IR Spectrum) |
| C-H: |
C-H: |
| 3780 |
3200 |
| 3805 |
3000 |
| 3819 |
2950 |
| 3827 |
2800 |
| 3847 |
2650 |
| Carbonyl: |
Carbonyl: |
| 2392 |
1730 |
| Aromatic Ring: |
Aromatic Ring: |
| 1929 |
2000 |
| 1905 |
1900 |
| O-H wave: |
O-H wave: |
| 1096 |
900 |
| 6-21 & 6-31 Transition energy (nm) |
6-21& 6-31 Oscillator
Strength |
| 202.6 |
0.001445 |
| 195.3 |
0.031782 |
| 189.6 |
0.072450 |
| 147.4 |
1.109531 |
| 144.6 |
0.833347 |
| 134.2 |
0.000008 |
| 131.3 |
0.000464 |
| 128.1 |
0.288010 |
| 126.3 |
0.000369 |
| 124.2 |
0.008856 |
Table 2: Shows the UV-Vis calculations using the 6-21 and 6-31 levels of theory. The data was compared to a UV-Vis spectrum from the NIST website. It was noticed that the calculated data fit into the same relative shape of the spectrum, however, the calculated excited energy states were too high. To see the NIST spectrum log on to: http://webbook.nist.gov/cgi/cbook.cgi?ID=C65850&Units=SI&Mask=400#UV-Vis-Spec
Conclusion:
The programs used in this experiment were useful in determining information that would be very difficult to calculate by hand. Computers also have the luxury of being able to use large basis sets and perform calculations on complex molecules. Unfortunately, data reported does not always provide very accurate results, which can be seen in our calculations above. Computer simulations are useful for student laboratory, but not reliable for industrial laboratory type settings.
References:
Gutow. "Molecular Orbitals Experiment". 9/05. Pg. 1
NIST website
Lide, D.R. The CRC Handbook of Chemistry and Physics. 81ed. New York: CRC Press, 2000.
Brooke_Cassidy/371_Protected/M01_Lab