MO calculations by Z. Chambers and Gage Kerscher
Introduction
Several
molecular orbital calculations were performed for hydrofluoric acid,
nitrogen dioxide, and 2-chlorobiphenyl. Additionally, electronic
transition energies were calculated for 2-chlorobiphenyl. The
calculations were preformed using various levels of theory:
semi-empirical (AM1 and PM3), and ab initio (6-21G, 6-31G, and
Double Zeta Valence). The Double Zeta Valence (DZV) yielded the
structural properties for all three molecules with the lowest energy
expectation values.
The following table lists the number of AO's and MO's found for each molecule using ab initio theory.

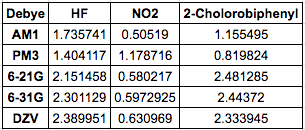
The next table contains experimentally derived dipole moments at each
level of theory. Experimental values obtained from NIST: HF - 1.820, NO2 - 0.316, 2-cholorobiphenyl - 1.43.
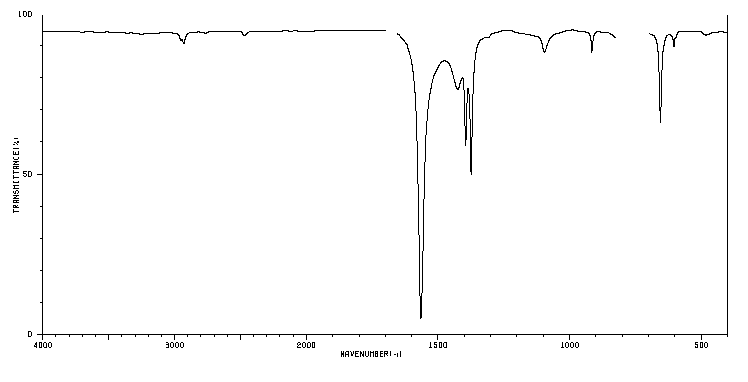
IR Spectra
NO2 (obtained from organicchem.org) The largest peak around 1580 inverse centimeters corresponds to symmetric stretching.
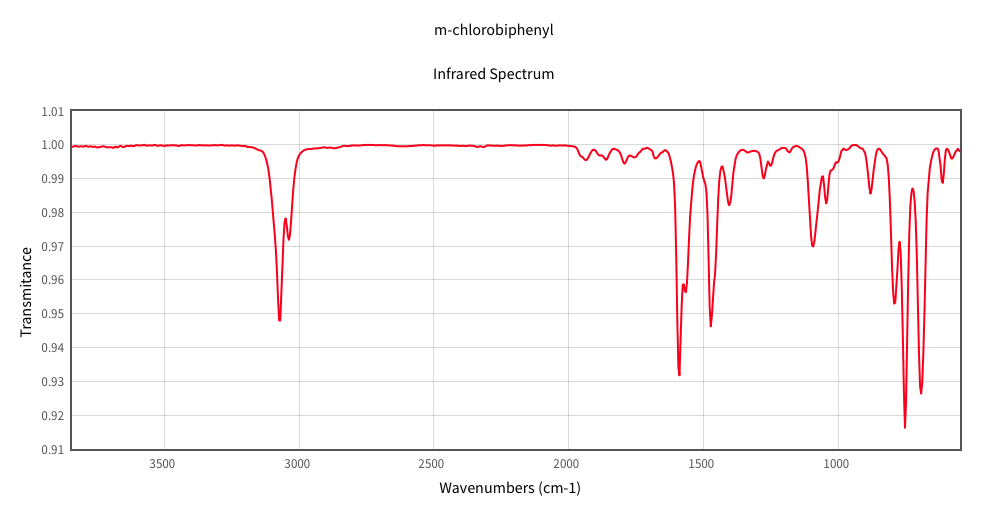
2-chlorobiphenyl (obtained from NIST). The peak near 1600 inverse
centimeters is due to C-C stretching and the one near C-H is due to C-H
stretching.
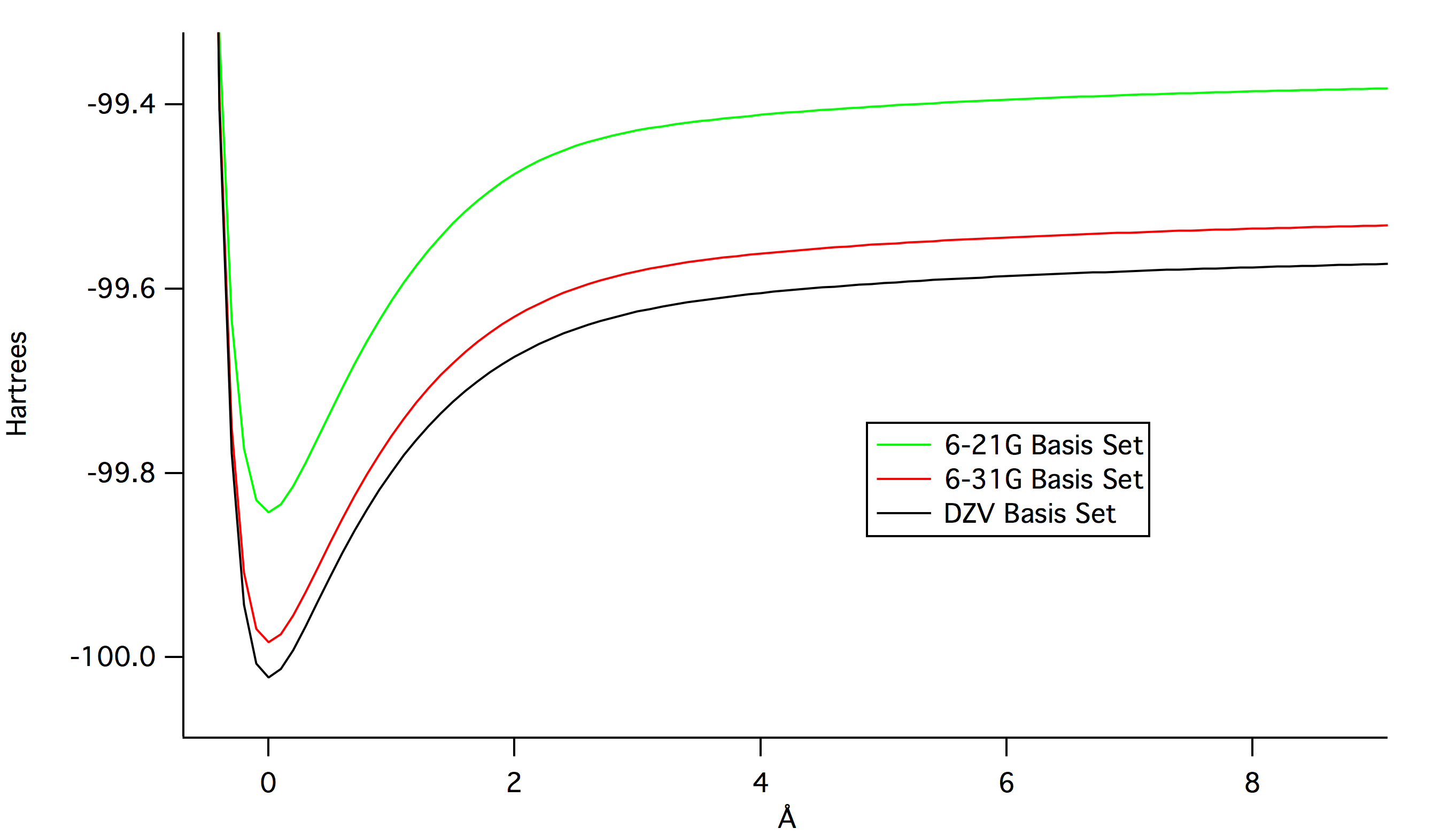
The following graphs illustrate the potential energy of bond stretching
for HF at different levels of theory. The first graph shows how far off
AM1, the smallest basis set was from the others. The second graph shows
that as the size of the basis set increases, the calculated potential
energy decreases.

Follow these links for results and discussion for each molecule:
Results on HF, NO2, and and 2-chlorobiphenyl.