Disodium
6-21G was the lowest level of theory used for geometry calculations.
The highest occupied molecular orbital (HOMO) is shown below. This is the orbital geometry that represents the valence electrons in their ground state. This was calculated by summing the total number of electrons for the molecule and dividing by two.
The lowest unoccupied molecular orbital (LUMO) is show below. This is the orbital that the valence electrons occupy once they have been excited by some form of energy. This was calculated by selecting the molecular orbital that was adjacent to the HOMO orbital but with a differing sign associated with the energy.
The electrostatic potential graphic shown below, represents the probability density of where the bonding electrons are located in a given molecule. Red colors represent regions of greater probability density, while blue colors represent regions of lower density. Colors between red and blue represent intermediate probability densities.
The partial atomic charges for disodium is shown in the graphic below. The atomic charges are 0 since disodium is composed of two of the same atom, so the charge separation is zero.
The different molecular bonding orbitals for disodium are shown in Table 1 below.
Table 1: Graphical representations of disodium's bonding and anti-bonding orbitals.
| Type of Bonding |
Graphical Representation |
| Sigma bonding |
 |
| Sigma anti-bonding |
 |
The potential energy graph, Figure 2, shown below graphs the total potential energy of disodium as the atoms move apart a given distance in nm. These potential energies were calculated at every level of theory. As the number of basis sets increased the calculated potential energy at each distance decreased. This is why the TZV calculation has the lowest potential energy curve.
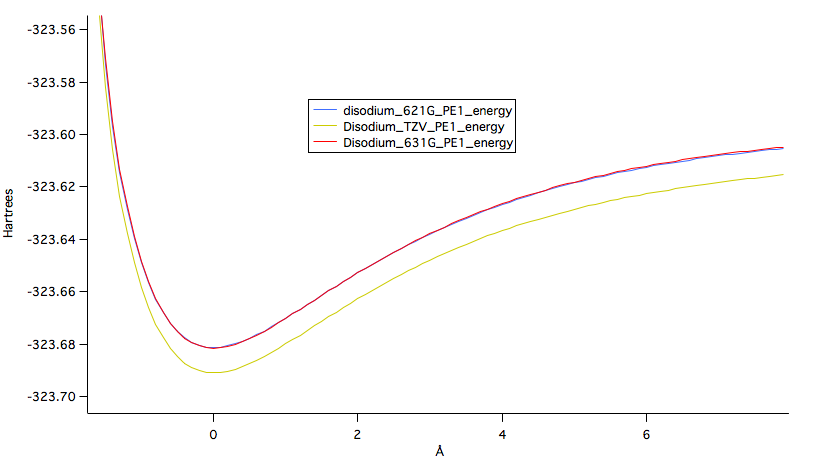
Figure 1: Potential energy curves generated at each level of theory.
As disodium is a symmetric molecule it does not absorb in the IR region. Because of this there are no vibrational frequencies to compared to. The TZV level of theory was able to calculate a single vibration at 153.84cm-1.
Additionally, since disodium is a symmetric molecule composed of the same atom there is no dipole moment. So there are no literature values to reference, nor were there any calculated values.
You may look at any of these intermediate views again by clicking on the appropriate button.
Page skeleton and JavaScript generated by export to web function using Jmol 14.2.12_2015.01.22 2015-01-22 21:48 on Mar 2, 2015.
This will be the viewer
If your browser/OS combination is Java capable, you will get snappier performance if you use Java