Once the molecule file is fully loaded, the image at right will become live. At that time the "activate 3-D" icon ![]() will disappear.
will disappear.
Cyclopropane
Using MacMol, five different levels of optimization were
compared: AM1, PM3, 6-21G, 6-31G, and DZV. It was found that the
DZV optimization gave the minimum energy making it the most
energetically favorable orientation. The literature values for
bond lengths is C-C: 1.50 angstroms and C-H: 1.08 angstroms. The
literature values for bond angles is H-C-H: 114.5 degrees, C-C-C: 60
degrees, and H-C-C: 117.9 degrees.2The optimization AM1 was further optimized using the 6-21G. The bond lengths and angles found in this optimization were the closest to the literature values making it the best of the three optimizations for bond lengths and angles.
The 6-21G was further optimized using the 6-31G.
The 6-31G was further optimized using the DZV. The DZV is the highest level of optimization in the ab initio. Even though this was further optimized, it did not yield values closer to the literature values than the 6-21G.
The HOMO is the highest occupied molecular orbital. This orbital is calculated by adding the electrons in each atom and then dividing by two. In the case of cyclopropane, there are 24 electrons, so the HOMO orbital is the 12th orbital.
The LUMO is the lowest unoccupied molecular orbital. This orbital is found by moving up to the next highest orbital from the HOMO. In this case, it was the 13th orbital. No electrons are normally here, however the electrons can be excited to the LUMO orbital.
The electrostatic potential is a representation of the electric potential energy as it varies throughout the molecule. In the image, the red area represents a higher density of the electrons and thus a higher electric potential energy. The blue area represents a lower density of electrons and thus a lower electric potential energy. The diagram shows that most of the electrons are being shared between the carbons because of the covalent bonds. Hydrogen only has one electron, so there is a smaller electron density in the hydrogen's proximity.
This is a non-polar molecule, therefore there are very small partial charges. Carbon has just a little higher electronegativity than hydrogen, but not enough to create large partial charges. The partial positive is on the hydrogen because it is lower in electronegativity.
The vibrational frequency for a C-H stretch is expected to be 3103cm^-1 according to literature.2 The experimental value for the C-H stretch was observed to be 3350cm^-1. This yields a percent error of 8%.
The vibrational frequency for a C-C stretch is expected to be 1188cm^-1 according to literature.2 The experimental value for the C-C stretch was observed to be 1206.6cm^-1. This yields a percent error of 2%.
The vibrational frequency for a C-H vibration is expected to be 854cm^-1 according to literature.2 The experimental value for the C-H vibration was observed to be 833.73cm^-1. This yields a percent error of 2%.
The vibrational frequency for a C-C vibration is expected to be 1479cm^-1 according to literature.2 The experimental value for the C-C vibration was observed to be 1328.8cm^-1. This yields a percent error of 10%.
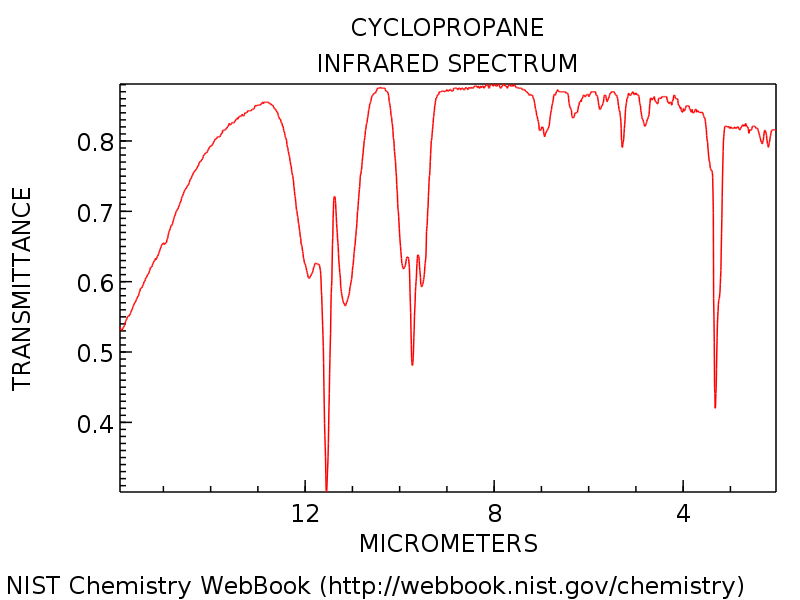
Figure 1: The infrared spectrum for cyclopropane.2
The vibrations shown above correspond with the IR spectrum found in literature.
The dipole moment in cyclopropane is 0 debeyes in literature, which makes sense because cyclopropane is non-polar. The experimental value was found to be 0.00005 debeyes using the DZV optimization.2
Page skeleton and JavaScript generated by export to web function using Jmol 14.2.12_2015.01.22 2015-01-22 21:48 on Mar 7, 2015.
This will be the viewer
If your browser/OS combination is Java capable, you will get snappier performance if you use Java