Molecular Orbital Calculations of Hydrogen Chloride
|
Jmol0 will appear here.
|
|
|
CLICK IMAGE TO ACTIVATE 3D
AM1 geometry optimization
|
|
AM1 geometry optimization gave a bond length value of 1.28 angstroms.
|
|
|
Jmol1 will appear here.
|
|
|
CLICK IMAGE TO ACTIVATE 3D
PM3 geometry optimization
|
|
PM3 geometry optimization gave a bond length value of 1.27 angstroms.
This proved to be the best level of theory for geometry optimization
because the value came closest to the literature value of 1.27
angstroms.7
|
|
|
Jmol2 will appear here.
|
|
|
CLICK IMAGE TO ACTIVATE 3D
DZV geometry optimization
|
|
DZV geometry optimization gave a bond length value of 1.26 angstroms.
DZV is the highest level of theory used for geometry optimization.
|
|
|
Jmol3 will appear here.
|
|
|
CLICK IMAGE TO ACTIVATE 3D
6-21G geometry optimization
|
|
6-21G geometry optimization gave a bond length value of 1.27 angstroms. This also proved to be the
best level of theory for geometry optimization because the value came
closest to the literature value of 1.27 angstroms.7 6-21G is the lowest
level of ab initio theory used for geometry optimization.
|
|
|
Jmol4 will appear here.
|
|
|
CLICK IMAGE TO ACTIVATE 3D
6-31G geometry optimization
|
|
6-31G geometry optimization gave a bond length value of 1.26 angstroms.
6-31G is the second highest level of ab initio theory for geometry optimization.
|
|
|
Jmol5 will appear here.
|
|
|
CLICK IMAGE TO ACTIVATE 3D
Highest occupied molecular orbital of HCl
|
|
This is the highest occupied molecular orbital (HOMO) at orbital 9.
Orbital 9 was chosen for the HOMO because HCl has a total number of 18
electrons, and the number of total electrons was divided by two.
|
|
|
Jmol6 will appear here.
|
|
|
CLICK IMAGE TO ACTIVATE 3D
Lowest unoccupied molecular orbital of HCl
|
|
This is the lowest unoccupied molecular orbital (LUMO) at orbital 10.
Orbital 10 was chosen as the LUMO because the HOMO is in orbital 9.
|
|
|
Jmol7 will appear here.
|
|
|
CLICK IMAGE TO ACTIVATE 3D
The electrostatic potential of HCl
|
|
This is the electrostatic potential of HCl. The red area represents the
lowest electrostatic potential and the blue area represents the highest
electrostatic potential. Intermediate colors represent intermediate
potentials.
|
|
|
Jmol8 will appear here.
|
|
|
CLICK IMAGE TO ACTIVATE 3D
Partial atomic charges on each atom of HCl
|
|
The partial atomic charge on each atom is shown in the diagram on the
right. The values of the partial charges on each atom were created by
the asymmetric distribution of electrons in the chemical bond.
|
|
|
Jmol9 will appear here.
|
|
|
CLICK IMAGE TO ACTIVATE 3D
Sigma bonding present in the H-Cl bond
|
|
The diagram on the left shows the sigma bonding orbital present in the
H-Cl bond as shown in figure 2. This is the lowest occupied energy
level.
|
|
|
Jmol10 will appear here.
|
|
|
CLICK IMAGE TO ACTIVATE 3D
Sigma-pi bonding present in the H-Cl bond
|
|
The diagram on the right shows the non-bonding orbital present in the H-Cl bond as shown in figure 2 below. This is the highest occupied energy orbital and is also degenerate.
|
|
|
Jmol11 will appear here.
|
|
|
CLICK IMAGE TO ACTIVATE 3D
Pi non-bonding orbital present in the H-Cl bond
|
|
The diagram on the left shows the pi non-bonding orbital present in the H-Cl bond as shown in figure 2 below. This is the highest occupied energy orbital and is also degenerate.
|
|
|
Jmol12 will appear here.
|
|
|
CLICK IMAGE TO ACTIVATE 3D
Vibrational stretch of HCl
|
|
The vibrational stretch of the HCl molecule can be visualized. The
calculated vibrational frequency associated with this motion is 3170.14
cm¯¹. The literature vibrational frequency 2990.10 cm¯¹.7
|
Figure 1 shows the potential
energy of the HCl bond as it is stretched and compressed.
Potential energy minima for all three basis sets occur at the ideal bond
length, where there is no stretching or compression. Figure 1 is a
direct comparison of the three ab initio basis sets that were
used. The lowest potential energy well is the most stable
configuration of the molecule, and therefore the most accurate
calculation. It is surprising that the largest basis set, DZV, is found
in the middle of the other two, as opposed to at the bottom. The lowest
potential energy actually comes from the mid-sized basis set, the
6-31G. This suggests that the size of the basis set is not the
only important parameter to consider when evaluating the validity of any
quantum calculations.
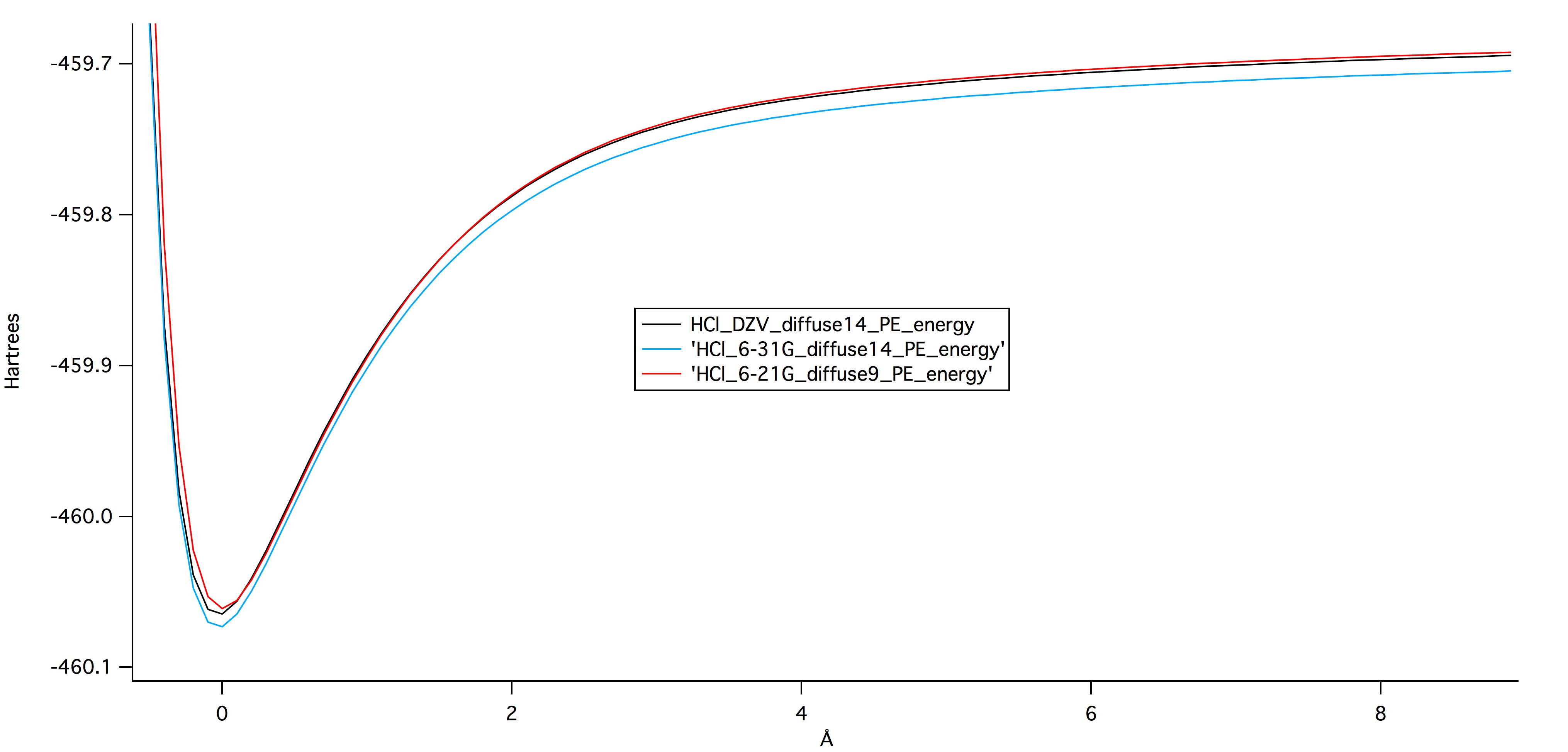 Figure 1:
Figure 1: Graph of potential energy vs bond length at different levels of theory. The graph was created in IGOR Pro.
|


Figure 2: molecular orbital diagram for hydrogen
chloride
Table 1: Calculated dipole moments
at different levels of theory and the percent error
(compared
literature value of 1.08.
7
The dipole moment was calculated at different levels of theory with the
inclusion of many different combinations of diffuse functions. The
values for "d", "f", and "light" functions were varied between 0 and 3
for each of the three
ab initio basis sets. Nine different
combinations were run in the 6-21G basis set with the best (closes to
literature value) dipole moment of 1.174649 D resulting from d=3, f=0,
light=3. Fourteen different combinations were run in the 6-31G
basis set with the
best dipole moment of 1.170069 D resulting from d=3, f=1,
light=3. Fourteen different combinations were also run in the DZV basis
set with the
best dipole moment of 1.153389 D resulting from d=3, f=1,
light=3. The DZV basis set plus diffuse functions produced the
closest to literature value for the dipole moment of 1.08 D
7, with a 6.80% margin of error. The best dipole calculations for each
ab initio basis set are summarized above in Table 1.
Based on template by A. Herráez as modified by J. Gutow
Using directory /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
adding JmolPopIn.js
...jmolApplet0
...adding HCl_AM1.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...copying
file:/Users/student/Desktop/Lab 2 Sean Boulanger/HCl/HCl_AM1.log
to
/Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb/HCl_AM1.log
...adding HCl_AM1.spt
...jmolApplet1
...adding HCl_PM3.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...copying
file:/Users/student/Desktop/Lab 2 Sean Boulanger/HCl/HCl_PM3.log
to
/Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb/HCl_PM3.log
...adding HCl_PM3.spt
...jmolApplet2
...adding HCl_DZV_diffuse14.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...copying
file:/Users/student/Desktop/Lab 2 Sean Boulanger/HCl/HCl_DZV_diffuse14.log
to
...compressing large data file to
/Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb/HCl_DZV_diffuse14.log.gz
/Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb/HCl_DZV_diffuse14.log.gz
...adding HCl_DZV_diffuse14.spt
...jmolApplet3
...adding HCl_6-21G_diffuse09.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...copying
file:/Users/student/Desktop/Lab 2 Sean Boulanger/HCl/HCl_6-21G_diffuse9.log
to
/Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb/HCl_6-21G_diffuse9.log
...adding HCl_6-21G_diffuse09.spt
...jmolApplet4
...adding HCl_6-31G_diffuse14.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...copying
file:/Users/student/Desktop/Lab 2 Sean Boulanger/HCl/HCl_6-31G_diffuse14.log
to
...compressing large data file to
/Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb/HCl_6-31G_diffuse14.log.gz
/Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb/HCl_6-31G_diffuse14.log.gz
...adding HCl_6-31G_diffuse14.spt
...jmolApplet5
...adding HCl_DZV_diffuse14_HOMO.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...adding HCl_DZV_diffuse14_HOMO.spt
...jmolApplet6
...adding HCl_DZV_diffuse14_LUMO.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...adding HCl_DZV_diffuse14_LUMO.spt
...jmolApplet7
...adding HCl_DZV_diffuse14_ElectrostaticPotential.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...adding HCl_DZV_diffuse14_ElectrostaticPotential.spt
...jmolApplet8
...adding HCl_DZV_diffuse14_PartialAtomicCharge.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...adding HCl_DZV_diffuse14_PartialAtomicCharge.spt
...jmolApplet9
...adding HCl_DZV_diffuse14_S_SigmaBonding.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...adding HCl_DZV_diffuse14_S_SigmaBonding.spt
...jmolApplet10
...adding HCl_DZV_diffuse14_SP_SigmaBonding.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...adding HCl_DZV_diffuse14_SP_SigmaBonding.spt
...jmolApplet11
...adding HCl_DZV_diffuse14_P_NonBonding.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...adding HCl_DZV_diffuse14_P_NonBonding.spt
...jmolApplet12
...adding HCl_DZV_diffuse14_VibrationalStretch.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...copying
file:/Users/student/Desktop/Lab 2 Sean Boulanger/HCl/HCl_DZV_vibfreq.log
to
...compressing large data file to
/Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb/HCl_DZV_vibfreq.log.gz
/Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb/HCl_DZV_vibfreq.log.gz
...adding HCl_DZV_diffuse14_VibrationalStretch.spt
...jmolApplet13
...adding SO2_AM1.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...copying
file:/Users/student/Desktop/Lab 2 Sean Boulanger/SO2/SO2_AM1.log
to
...compressing large data file to
/Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb/SO2_AM1.log.gz
/Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb/SO2_AM1.log.gz
...adding SO2_AM1.spt
...jmolApplet14
...adding SO2_PM3.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...copying
file:/Users/student/Desktop/Lab 2 Sean Boulanger/SO2/SO2_PM3.log
to
...compressing large data file to
/Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb/SO2_PM3.log.gz
/Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb/SO2_PM3.log.gz
...adding SO2_PM3.spt
...jmolApplet15
...adding SO2_6-21G.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...copying
file:/Users/student/Desktop/Lab 2 Sean Boulanger/SO2/SO2_6-21G.log
to
/Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb/SO2_6-21G.log
...adding SO2_6-21G.spt
...jmolApplet16
...adding SO2_6-31G.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...copying
file:/Users/student/Desktop/Lab 2 Sean Boulanger/SO2/SO2_6-31G.log
to
/Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb/SO2_6-31G.log
...adding SO2_6-31G.spt
...jmolApplet17
...adding SO2_DZV.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...copying
file:/Users/student/Desktop/Lab 2 Sean Boulanger/SO2/SO2_DZV.log
to
/Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb/SO2_DZV.log
...adding SO2_DZV.spt
...jmolApplet18
...adding SO2_ElectrostaticPotential.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...adding SO2_ElectrostaticPotential.spt
...jmolApplet19
...adding SO2_PartialAtomicCharge.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...adding SO2_PartialAtomicCharge.spt
...jmolApplet20
...adding SO2_DZV_HOMO.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...adding SO2_DZV_HOMO.spt
...jmolApplet21
...adding SO2_DZV_LUMO.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...adding SO2_DZV_LUMO.spt
...jmolApplet22
...adding SO2_DZV_VibrationBend.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...copying
file:/Users/student/Desktop/Lab 2 Sean Boulanger/SO2/SO2_DZV_vibfreq.log
to
...compressing large data file to
/Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb/SO2_DZV_vibfreq.log.gz
/Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb/SO2_DZV_vibfreq.log.gz
...adding SO2_DZV_VibrationBend.spt
...jmolApplet23
...adding SO2_DZV_VibrationStretch.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...adding SO2_DZV_VibrationStretch.spt
...jmolApplet24
...adding SO2_DZV_VibrationSimStretch.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...adding SO2_DZV_VibrationSimStretch.spt
...jmolApplet25
...adding mdClb_AM1.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...copying
file:/Users/student/Desktop/Lab 2 Sean Boulanger/m-dichlorobenzene/mdClb_AM1.log
to
...compressing large data file to
/Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb/mdClb_AM1.log.gz
/Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb/mdClb_AM1.log.gz
...adding mdClb_AM1.spt
...jmolApplet26
...adding mdClb_PM3.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...adding mdClb_PM3.spt
...jmolApplet27
...adding mdClb_6-21G.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...copying
file:/Users/student/Desktop/Lab 2 Sean Boulanger/m-dichlorobenzene/mdClb_6-21G.log
to
...compressing large data file to
/Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb/mdClb_6-21G.log.gz
/Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb/mdClb_6-21G.log.gz
...adding mdClb_6-21G.spt
...jmolApplet28
...adding mdClb_6-31G.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...copying
file:/Users/student/Desktop/Lab 2 Sean Boulanger/m-dichlorobenzene/mdClb_6-31G.log
to
...compressing large data file to
/Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb/mdClb_6-31G.log.gz
/Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb/mdClb_6-31G.log.gz
...adding mdClb_6-31G.spt
...jmolApplet29
...adding mdClb_DZV.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...copying
file:/Users/student/Desktop/Lab 2 Sean Boulanger/m-dichlorobenzene/mdClb_DZV.log
to
...compressing large data file to
/Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb/mdClb_DZV.log.gz
/Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb/mdClb_DZV.log.gz
...adding mdClb_DZV.spt
...jmolApplet30
...adding mdClb_DZV_HOMO.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...adding mdClb_DZV_HOMO.spt
...jmolApplet31
...adding mdClb_DZV_LUMO.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...adding mdClb_DZV_LUMO.spt
...jmolApplet32
...adding mdClb_DZV_ElectroStaticPotential.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...adding mdClb_DZV_ElectroStaticPotential.spt
...jmolApplet33
...adding mdClb_DZV_PartialAtomicCharge.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...adding mdClb_DZV_PartialAtomicCharge.spt
...jmolApplet34
...adding mdClb_DZV_Hstretch.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...copying
file:/Users/student/Desktop/Lab 2 Sean Boulanger/m-dichlorobenzene/mdClb_DZV_vibfreq.log
to
...compressing large data file to
/Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb/mdClb_DZV_vibfreq.log.gz
/Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb/mdClb_DZV_vibfreq.log.gz
...adding mdClb_DZV_Hstretch.spt
...jmolApplet35
...adding mdClb_DZV_Hwiggle.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...adding mdClb_DZV_Hwiggle.spt
...jmolApplet36
...adding mdClb_DZV_Cwobble.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...adding mdClb_DZV_Cwobble.spt
...jmolApplet37
...adding mdClb_DZV_Hjiggle.png
copying and unzipping jsmol.zip directory into /Users/student/Desktop/Lab 2 Sean Boulanger/SBEK/HCl_SO2_mdClb
...adding mdClb_DZV_Hjiggle.spt