Once the molecule file is fully loaded, the image at right will become live. At that time the "activate 3-D" icon will disappear.
Hydrogen Cyanide (HCN)
The geometry optimization of 621G, 631G, and DZV are
shown below for the smallest polar molecule hydrogen cyanide. There is no bond
angle due to the molecule being linear making the angle 180 degrees.
Table 1: This table represents the bond lengths of each atom in hydrogen cyanide taken from the literature3.
Type of bond
Bond Length in Angstroms
C-H
1.064
C-N
1.156
These buttons below display the bond lengths calculated from each level of theory.
This button displays the bond length using the lowest level of optimization theory.
This button displays the bond length using the second highest level of optimization theory.
This button displays the bond length using the
highest level of theory and gave the best optimized lengths compared to
the literature. This comparison gave a 0.90% error for the H to N bond
length (Total Length of HCN).
This displays the Highest Occupied Molecular Orbital
found at orbital 7. The orbitals were calculated by adding all of
electrons in the molecule and dividing it by two.
This displays the Lowest Unoccupied Molecular Orbital
found at orbital 8. The electrons will transition to this orbital when
excited with enough energy.
This button displays the electrostatic potential. The
electron cloud ranges in color from red to blue. Red being areas of
high electrostatic potential and blue being areas of low
electrostatic potential. Since the nitrogen atom is more
electronegative, thus pulling the electrons closer to it (Red area).
The partial atomic charge on each atom is shown in
this diagram. They are created by the asymmetric distribution of
electrons in a chemical bond. The sum of these charges equal to zero.
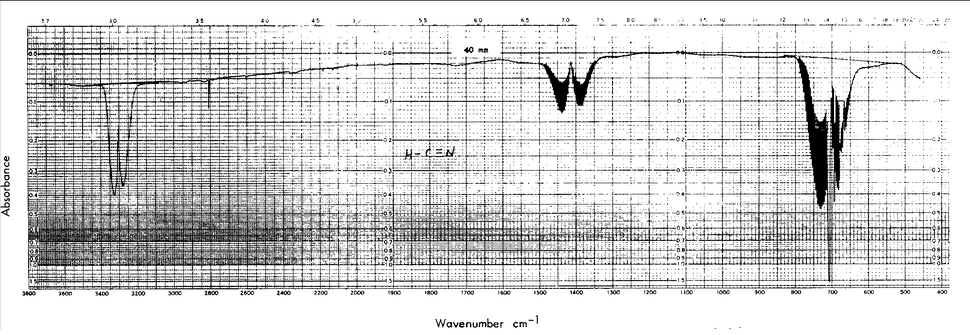
Below shows the vibrations of HCN, determined from an IR-spectrum.
Table 2: The vibrations of HCN in cm^-1 taken from the literature1.
Type of vibration
Wave Length in cm^-1
C-H bend
685-995
C-N triple bond stretch
2200-2280
C-H alkyne stretch
3200-3310
Figure 1:
An IR-Spectrum of HCN taken from the NIST website3.
C-H bend
C-N triple bond stretch
C-H alkyne stretch
These buttons above represent the vibrations
calculated from the highest level of theory, DZV. The values calculated
from the DZV optimization does not deviate with the C-H bend, but with
the C-H triple bond stretch and the C-H alkyne stretch it deviates
substantially. The values for those two vibrations were overestimated
giving a 2.16% and 12.20% error respectively.
A Dipole moment is present in HCN since the molecule is polar. Table 3: This table displays the dipole moment for HCN in the gas phase taken from the literature5.
Molecule
Dipole Moment
Hydrogen Cyanide
3.06
Table 4: This table displays the dipole moments calculated from each level of theory.
Level of Geometry Optimization Theory
Dipole Moment of HCN
621G
3.028709
631G
3.271937
DZV
3.286865
The best value for the dipole moment of HCN was
calculated with the lowest level of theory, 621G. This gave a percent
error of 1.04%.
Based on template by A. Herráez as modified by J. Gutow
Using directory /Volumes/ZAC'S DRIVE/Spring 2015/Pchem Website Ryan and Larry/HCN page
adding JmolPopIn.js
...jmolApplet0
...adding Bond_Length_621G_HCN.png
copying and unzipping jsmol.zip directory into /Volumes/ZAC'S DRIVE/Spring 2015/Pchem Website Ryan and Larry/HCN page
...copying
file:/Volumes/ZAC'S DRIVE/Lab 2 QM Calculations /HCN_621G_opt.log
to
/Volumes/ZAC'S DRIVE/Spring 2015/Pchem Website Ryan and Larry/HCN page/HCN_621G_opt.log
...adding Bond_Length_621G_HCN.spt
...jmolApplet1
...adding Bond_Length_631G_HCN.png
copying and unzipping jsmol.zip directory into /Volumes/ZAC'S DRIVE/Spring 2015/Pchem Website Ryan and Larry/HCN page
...copying
file:/Volumes/ZAC'S DRIVE/Lab 2 QM Calculations /HCN_631G_opt.log
to
/Volumes/ZAC'S DRIVE/Spring 2015/Pchem Website Ryan and Larry/HCN page/HCN_631G_opt.log
...adding Bond_Length_631G_HCN.spt
...jmolApplet2
...adding Bond_Length_DZV_HCN.png
copying and unzipping jsmol.zip directory into /Volumes/ZAC'S DRIVE/Spring 2015/Pchem Website Ryan and Larry/HCN page
...copying
file:/Volumes/ZAC'S DRIVE/Lab 2 QM Calculations /HCN_DZV_opt.log
to
/Volumes/ZAC'S DRIVE/Spring 2015/Pchem Website Ryan and Larry/HCN page/HCN_DZV_opt.log
...adding Bond_Length_DZV_HCN.spt
...jmolApplet3
...adding HOMO_HCN.png
copying and unzipping jsmol.zip directory into /Volumes/ZAC'S DRIVE/Spring 2015/Pchem Website Ryan and Larry/HCN page
...adding HOMO_HCN.spt
...jmolApplet4
...adding LUMO_HCN.png
copying and unzipping jsmol.zip directory into /Volumes/ZAC'S DRIVE/Spring 2015/Pchem Website Ryan and Larry/HCN page
...adding LUMO_HCN.spt
...jmolApplet5
...adding Electrostatic_Potential_HCN.png
copying and unzipping jsmol.zip directory into /Volumes/ZAC'S DRIVE/Spring 2015/Pchem Website Ryan and Larry/HCN page
...adding Electrostatic_Potential_HCN.spt
...jmolApplet6
...adding Partial_Atomic_Charge_HCN.png
copying and unzipping jsmol.zip directory into /Volumes/ZAC'S DRIVE/Spring 2015/Pchem Website Ryan and Larry/HCN page
...adding Partial_Atomic_Charge_HCN.spt
...jmolApplet7
...adding 2329_23_cm_-1_CN.png
copying and unzipping jsmol.zip directory into /Volumes/ZAC'S DRIVE/Spring 2015/Pchem Website Ryan and Larry/HCN page
...copying
file:/Volumes/ZAC'S DRIVE/Lab 2 QM Calculations /HCN_Vib_DZV.log
to
...compressing large data file to
/Volumes/ZAC'S DRIVE/Spring 2015/Pchem Website Ryan and Larry/HCN page/HCN_Vib_DZV.log.gz
/Volumes/ZAC'S DRIVE/Spring 2015/Pchem Website Ryan and Larry/HCN page/HCN_Vib_DZV.log.gz
...adding 2329_23_cm_-1_CN.spt
...jmolApplet8
...adding 3713_98_cm_-1_C-H_.png
copying and unzipping jsmol.zip directory into /Volumes/ZAC'S DRIVE/Spring 2015/Pchem Website Ryan and Larry/HCN page
...adding 3713_98_cm_-1_C-H_.spt
...jmolApplet9
...adding 886_81_cm_-1_C-H_bend.png
copying and unzipping jsmol.zip directory into /Volumes/ZAC'S DRIVE/Spring 2015/Pchem Website Ryan and Larry/HCN page
...adding 886_81_cm_-1_C-H_bend.spt
This will be the viewer
If your browser/OS combination is Java capable, you will get snappier performance if you use Java