Hydrogen Fluoride (HF)
The model for the best geometry optimization is shown in a live display using the Double Zeta Valence level of theory. The DZV level of theory was best, because it used the largest basis set size. The geometry results from all the levels of ab initio theory are compared to experimental values shown in Table 1.
|
|
| Table 1: HF Bond
Lengths |
||
| Theory |
Length (nm) |
|
| AM1 |
0.099 nm |
|
| 621-G |
0.083 nm |
|
| 631-G |
0.083 nm |
|
| Experimental |
0.0917 1 |
|
The HOMO or highest occupied molecular orbital is shown in a live display using the DZV level of theory.
|
|
The LUMO or the lowest occupied molecular orbital is shown in a live display using the DZV level of theory.
|
|
The molecular electrostatic potential is shown using the DZV level of theory. This live display shows the electrostatic potential at a certain point of the molecule with red meaning a negative potential and blue meaning a positive potential.
|
|
The partial atomic charges are shown in a live display using the DZV level of theory with the corresponding value on each atom.
|
|
The dipole moments were calculated using different levels of ab initio theory compared to the literature value. The 6-31G with two D functions, one F function, and two light functions calculated the most accurate dipole strength.
| Table 2: Dipole
Moments |
|
| Theory: (D functions, F functions, light functions) |
Dipole Strength (Debye) |
| AMI (0,0,0) |
1.735716 |
| 621-G (0,0,0) |
2.151451 |
| 631-G (0,0,0) |
2.301124 |
| DZV (0,0,0) |
2.389922 |
| 6-21G (1,0,1) |
1.993306 |
| 6-31G (2,1,2) |
1.835002 |
| DZV (2,1,2) |
1.897335 |
| Experimental |
1.820 1 |
| Table 3: Energy
Level Diagram |
|
| Energy (a.u.) Bond Type |
Orbital Diagram (DZV) |
| E = 1.9631 anti-bonding |
 |
| E = 1.158 anti-bonding |
 |
| E = 1.0963 anti-bonding |
 |
| E = 1.0576 anti-bonding |
 |
| E = 1.0576 anti-bonding |
 |
| E = 0.1989 anti-bonding |
 |
| E = -0.6427 anti-bonding |
 |
| E = -0.6427 anti-bonding |
 |
| E = -0.7521 anti-bonding |
 |
| E = -1.6009 bonding |
 |
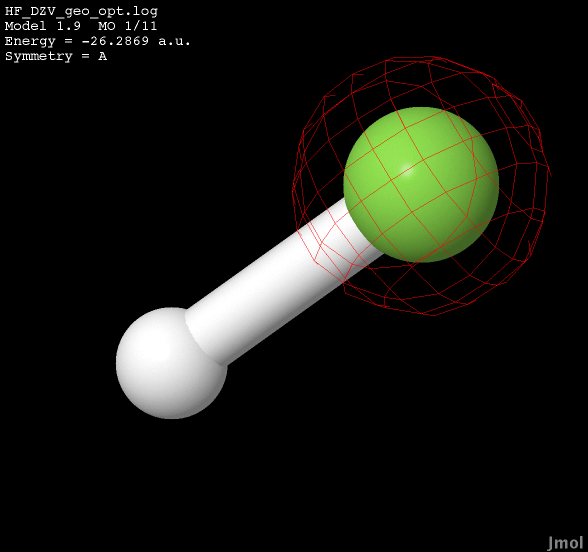
| E = -26.2869 non-bonding |
 |
A plot of the potential energy of bond stretching at three levels of ab initio theory is shown in Figure 1. The DZV level of theory calculated the lowest potential energy. DZV uses the biggest basis set size making a more accurate calculation compared to 621-G and 631-G.
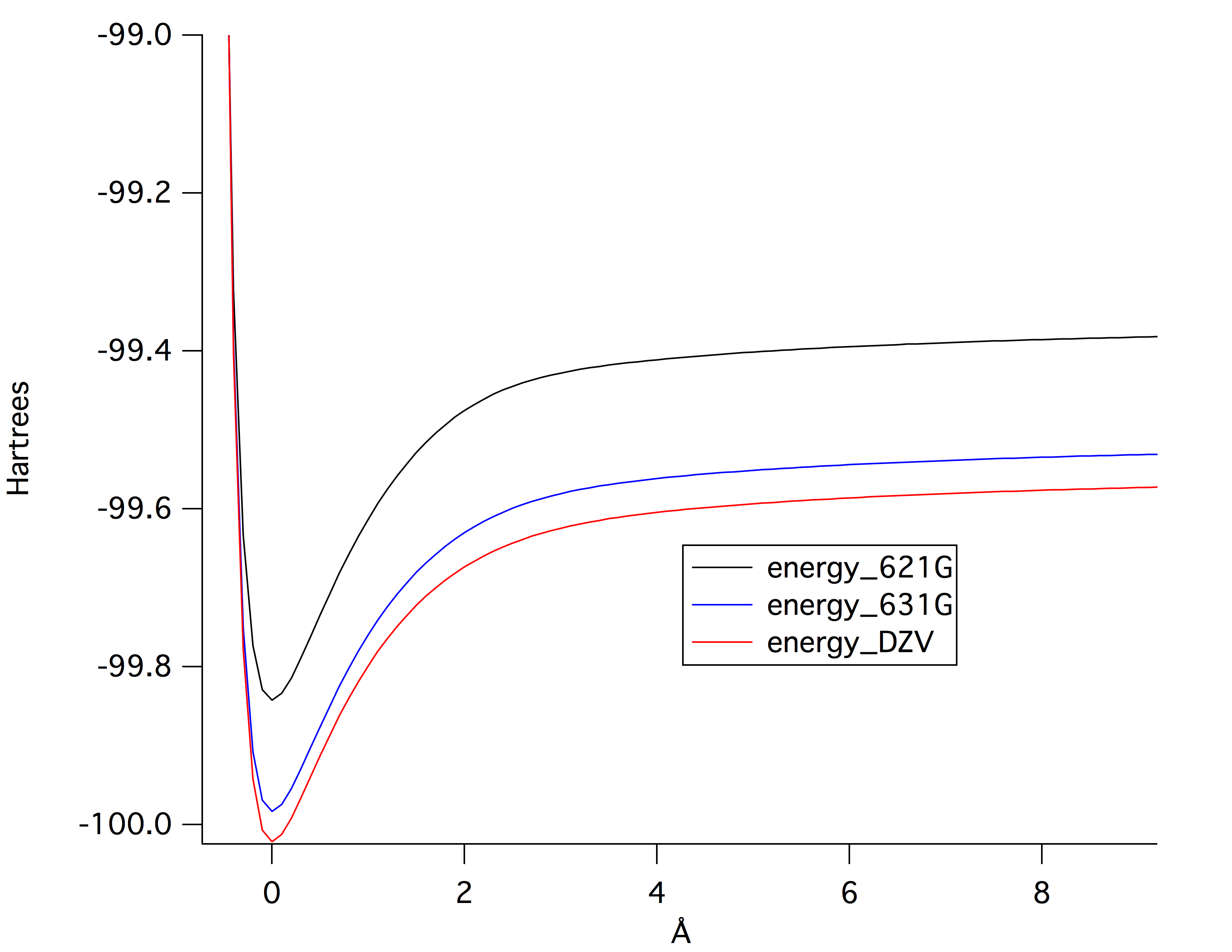
Figure 1: potential energy of bond stretching at three levels of ab initio
The calculated vibration energy was 4234.2603 cm^-1. More details can be seen by going to the Vibrational Calculations page.
NFH2
Anisole
Vibrational Calculations
Home Page
References
1 http://cccbdb.nist.gov/exp2.asp#NISTdiatomic
Page
skeleton and JavaScript generated by export to web
function using Jmol
12.2.34 2012-08-09 20:37 on Feb 25, 2013.