Hydrogen Cyanide Quantum Calculations
All of the live displays on this page were created using the best optimized geometry. The best found geometry optimization was at the ab initio theory level of 6-21G.
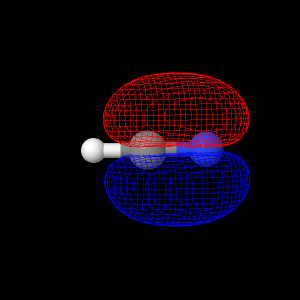
Hydrogen Cyanide has doubly degenerate HOMO orbitals. The
button below shows the first HOMO orbital for hydrogen cyanide.
All of the live displays on this page were created using the best optimized geometry. The best found geometry optimization was at the ab initio theory level of 6-21G.
|
|
The button below shows the second HOMO orbital for Hydrogen Cyanide.
|
|
Clicking the button below will show a live display of one of the LUMO orbitals.
|
|
The button below will show the display of the second LUMO orbital.
|
|
The bond length for the H-C bond was calculated to be 1.05Å, which was lower than the literature value of 1.064Å.1 The error in the bond length is 1.3%.
The C-N bond length was 1.14Å, and when compared to the literature value of 1.156Å, yields an error of 1.4%.1
|
|
The bond angle calculated was 180 degrees, which was expected due to the linear orientation of the molecule.
|
|
All bond lengths were the same for the different basis sets. As for the dipole moments, 6-21G showed a value closest to the reference value of 2.980 D; this is why this basis set was chosen.1
|
|
The table below shows the dipole moments of four basis sets. Diffuse functions were added to the basis sets in an attempt to improve the dipole moment. In the table, #D refers to the number of D heavy atom polarization functions, #F refers to the number of F heavy atom polarization functions, and #Light refers to the number of light atom polarization functions.
| Basis Set |
#D |
#F |
#Light |
Dipole (Debye) |
| AM1 |
0 |
0 |
0 |
2.361377 |
| 6-21G |
0 |
0 |
0 |
3.028704 |
| 6-21G | 1 |
0 |
1 |
3.020112 |
| 6-21G | 2 |
0 |
2 |
3.047205 |
| 6-31G | 0 |
0 |
0 |
3.241412 |
| 6-31G | 1 |
0 |
1 |
3.197838 |
| 6-31G | 2 |
0 |
2 |
3.173012 |
| 6-31G | 2 |
1 |
2 |
Error |
| 6-31G | 3 |
0 |
3 |
3.235740 |
| DZV |
0 |
0 |
0 |
3.286947 |
| DZV | 1 |
0 |
1 |
3.205086 |
| DZV | 2 |
1 |
2 |
Error |
| DZV | 2 |
0 |
2 |
3.187133 |
| DZV | 3 |
0 |
3 |
3.213797 |
The button below shows the partial atomic charges for hydrogen cyanide.
|
|
The button below shows the electrostatic potential for hydrogen cyanide.
|
|
The energy of the 1.6 vibration is 886.74 cm-1.
|
|
The energy for the 1.7 vibration is 886.74 cm-1.
|
|
The vibration energy associated with vibration 1.8 is 2329.3 cm-1. This is caused by symmetrical stretching in both the C-H and C-N bonds.
|
|
Vibration 1.9 has an energy of 3714.1401 cm-1. This is caused mainly by the C-H stretch but does have a contribution for the C-N stretch as well.
|
|
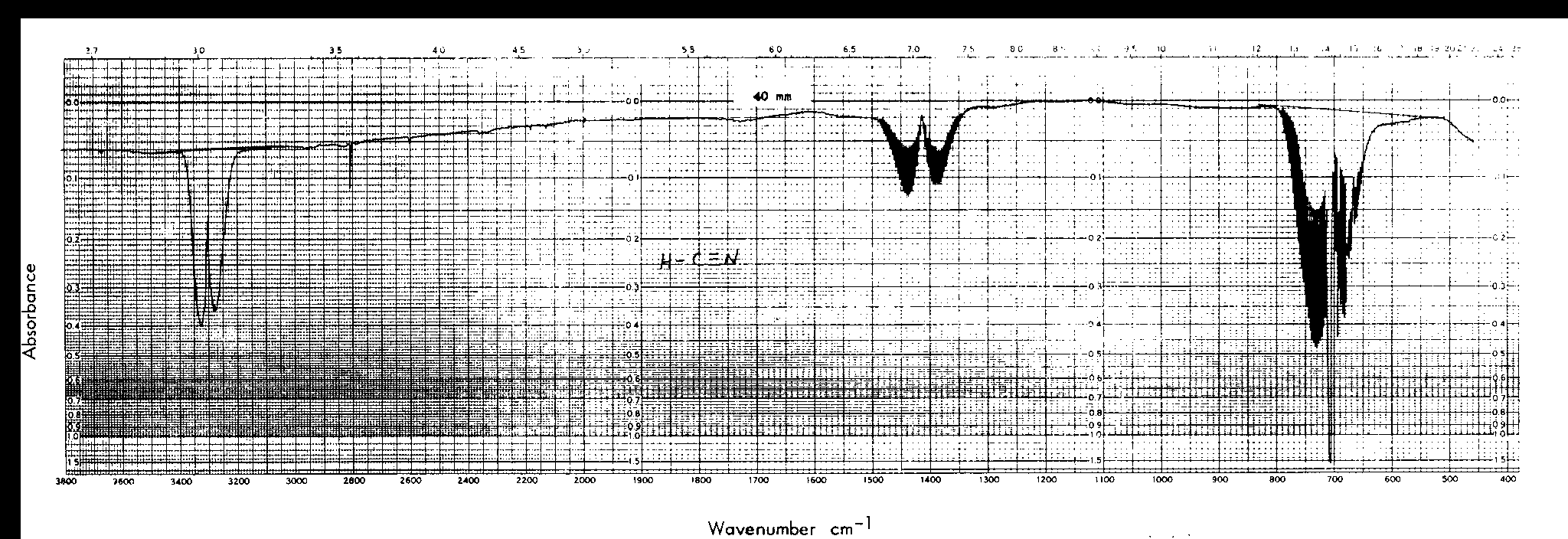
This IR was found on the NIST website.1
References
Page
skeleton and JavaScript generated by export to web function
using Jmol 12.2.34
2012-08-09 20:37 on Feb 26, 2013.