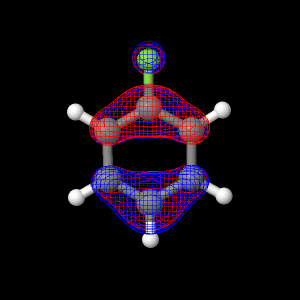
All of the live displays on this page were created using the best optimized geometry. The best found geometry optimization was at the ab initio theory level of DZV.
The energy of the HOMO orbital is -0.3536 a.u.
The button below shows the HOMO orbital for fluorobenzene
|
|
The LUMO orbital of fluorobenzene is found at an energy of 0.1069 a.u.
Clicking the button below will show a live display of the LUMO orbital.
|
|
Clicking on the button below will display a live model of fluorobenzene with bond lengths.
|
|
The two C-C bonds closest to the fluorine have a length of 1.38 Å, which is lower than the reference value of 1.392 Å.1 The error percent of these bonds is 1.2%.
The remaining 4 C-C bonds have lengths of 1.4 Å. Comparing this to the literature value of 1.392 Å yields an error of 0.83%. 1
The C-H bonds in the molecule have bond lengths of 1.07 Å. The error is 1.1% when compared to the literature value of 1.082 Å.1
The button below shows bond angles of fluorobenzene.
|
|
The DZV basis set was chosen as the best for this molecule because it is the largest of the tested basis sets. The DZV basis set, being larger, hopefully accounts for everything in this complex molecule.
|
|
The button below shows the partial atomic charges for fluorobenzene.
|
|
The button below shows the electrostatic potential for fluorobenzene.
|
|
The 8 main contributions to the energy vibrations are discussed and shown below.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
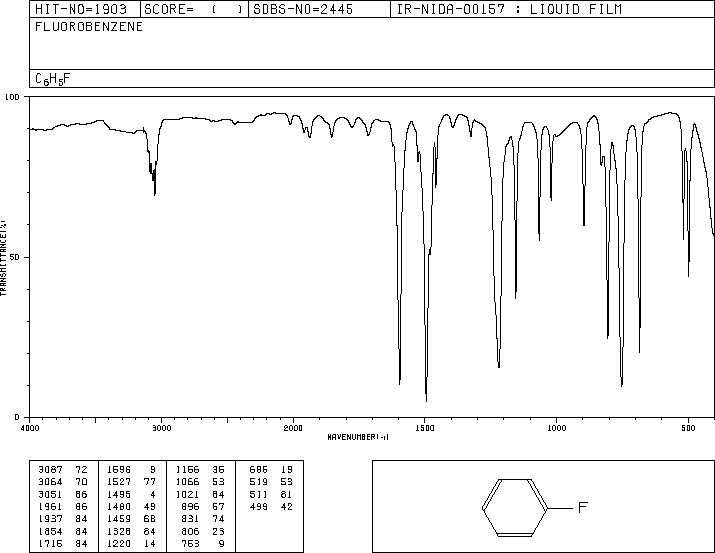
The IR spectrum for fluorobenzene below was found on the website for the spectral database system for organic compounds.2
The dipole moment for fluorobenzene was found to be 1.575356 Debye and when compared to the literature value of 1.66 Debye, has an error of 5.1%.1 This was done using an AM1 basis set, which is a very small basis set in comparison to some of the other basis sets. The DZV basis set, which is the largest, actually had the worst dipole moment value out of all of them.
Below is a table of calculated excitation energies for UV-Vis spectroscopy of Fluorobenzene.
| Basis Set |
Excitation Energy (nm) | Oscillation Strength |
Excited State |
| 6-21G |
186.02 |
0.017457 |
1 |
| 6-21G | 138.38 |
1.108524 |
3 |
| 6-21G | 136.98 |
1.150438 |
4 |
| 6-21G | 124.79 |
0.009360 |
6 |
| 6-21G | 114.75 |
0.001383 |
10 |
| 6-31G |
189.64 |
0.012066 |
1 |
| 6-31G | 182.20 |
0.001278 |
2 |
| 6-31G | 141.08 |
1.119436 |
3 |
| 6-31G | 139.75 |
1.194338 |
4 |
| 6-31G | 129.69 |
0.003585 |
5 |
| 6-31G | 125.33 |
0.009131 |
7 |
| DZV |
194.23 |
0.011203 |
1 |
| DZV | 187.01 |
0.002473 |
2 |
| DZV | 144.86 |
1.194734 |
3 |
| DZV | 143.94 |
1.271145 |
4 |
| DZV | 127.09 |
0.010794 |
5 |
| DZV | 124.90 |
0.020913 |
7 |
| DZV | 117.83 |
0.005455 |
9 |
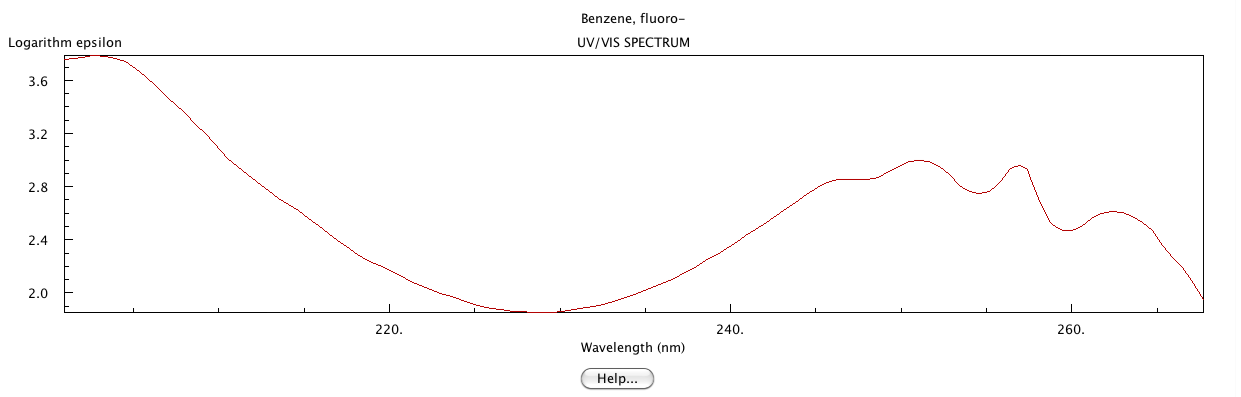
The following UV-Vis spectrum was obtained from the NIST.1
References
Page
skeleton and JavaScript generated by export to web function
using Jmol 12.2.34
2012-08-09 20:37 on Feb 26, 2013.