Trifluoromethane, and Chlorobenzene
By: Jesse Klein and Carly Salter
Abstract:
Through computational chemistry the geometry of a
molecule can be found by applying classical an quantum
mechanics to the molecule and adjusting the geometry of the
molecule until the lowest possible energy of the molecule is
found. This optimized molecule can then be subjected
to various symbolic experiments to give results which come
close to or even match those seen experimentally in the
laboratory and in nature.
Introduction:
The geometry of a given molecule effects that molecule's
physical and chemical properties. Through
computational chemistry, the geometry of a molecule can be
optimized, and from this optimization, the physical and
chemical properties seen experimentally can be seen
computationally. This geometry optimization is
achieved through determining the wavefunctions of electrons
and thereby determining the probability of an electron being
in a given location within the molecule. The issue
with this determination is that in a many electron molecule
(most molecules) the wavefunction can only be known for a
single electron. Only a guess can be made as to the
wavefunctions of all the other electrons in the
molecule. What is known is that the lower energy state
is always the preferred one in a molecule. With this
in mind, approximations of the wavefunctions of all the
other electrons while fixing the wavefunction for one
electron in the molecule can be made. Using geometry
generating software, such as MacMolPlt or Jmol, and
computational software, such as GamessQ, the geometry of a
given molecule can be determined by adjusting the
wavefunctions of the electrons and the geometry of the
molecule to find the lowest possible energy of the
molecule. This "tweaking" of the molecule is achieved
by fixing the wavefunction of one electron and guessing the
wavefunctions of the other electrons. The molecule's
geometry is then "tweaked" until the lowest energy is found
with a specific molecule geometry. Then a different
electron's wavefunction is fixed and the process begins
again. This process is repeated until the energy of
the molecule is brought down to the lowest possible energy
which goes with it a specific molecular geometry. From
this geometry a countless number of physical and chemical
properties can be found.
Using Jmol software, the geometry optimized molecule can
be put through a series of symbolic tests to see how the
molecule reacts under certain conditions. Since the
physics is already understood about such physical properties
as infrared spectroscopy, bond length, bond angle, etc. the
mathematical calculations which give the physical properties
desired can be applied to the geometry optimized
molecule. In many cases, these results have matched
results seen experimentally using physical samples of the
molecules in question.
Geometry optimization calculations can be time consuming
and expensive. To speed up this process, the physics
which governs the geometry optimization is applied in
"chunks" so to speak. Classical physics is first
applied to the molecule and the geometry optimized using
these parameters of physics. This is then followed by
slowly working in all the quantum mechanics needed over a
series of a few additional optimizations. In each
optimization, a better model using higher theory is applied
to the previous level of theory. By gradually changing
the models used, the compute time for finding the geometry
optimization of a given molecule is reduced.
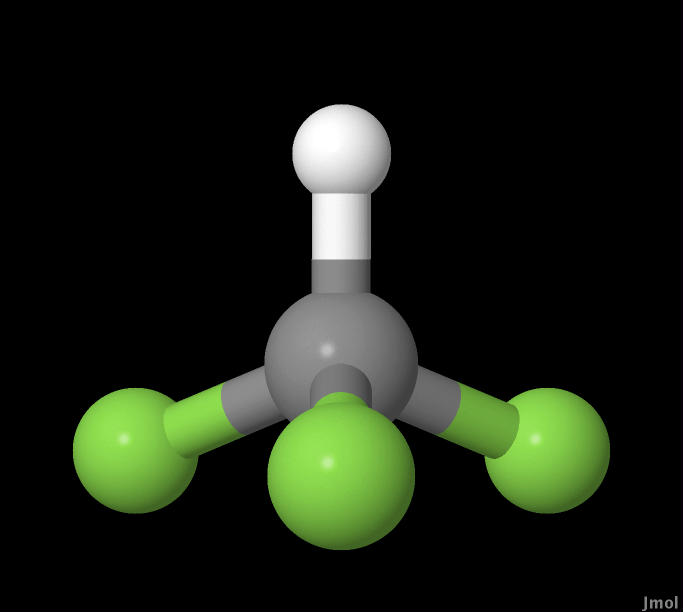
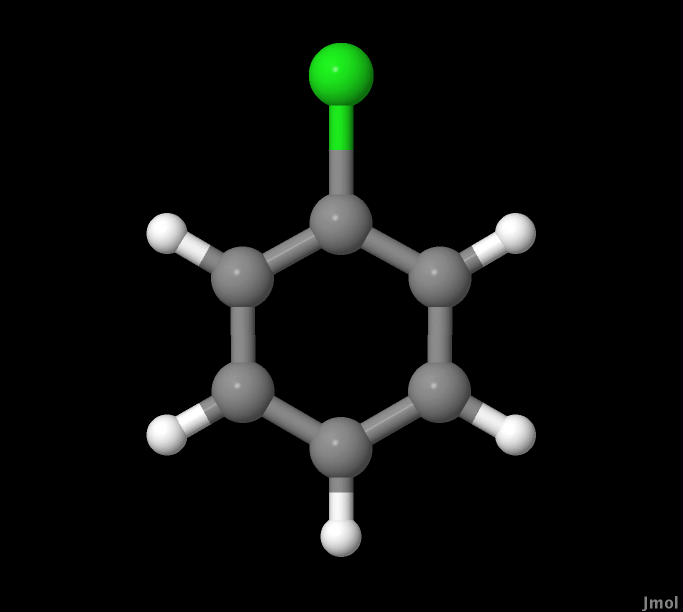
In this experiment CHF3, HBr, and C6H5Cl molecules were
optimized and the physical properties tested and compared
with experimental results. Theory levels of 3-21G (or
6-21G), 6-31G, and DZV were applied one after the other
until a geometry was found for each molecule which gave the
lowest possible energy. The results of the geometry
optimization and the physical and chemical property tests
performed can be seen for each molecule by following the
links below.
 |
 |
 |
| HBr Molecular Orbital
Link |
CHF3
Molecular Orbital Link |
C6H5Cl
Molecular Orbital Link |
Conclusion:
Given the level of theory used it is not surprising that
many of the physical properties found came incredibly close
to those found in literature. A further optimization
taking into account geometries which include unpaired
electrons could give a geometry with a lower energy level
but given the results found the geometries found is pretty
good approximations. Additionally, the Huckel
approximation was used in these computations which takes
into account only interactions between neighboring
atoms. By removing this approximation, the computing
time would increase but the results would be closer to those
found in literature.
References