Once the molecule file is fully loaded the image at right will become live. At that time the "activate 3-D" icon
![]() will disappear.
will disappear.
Nitric Oxide
The geometry optimizations for the three
highest levels of theory are shown below. The bond angle is not
included because it is 180 degrees. The literature3 value of 1.0619
angstroms for bond length was most closely found by the 6-31G level of theory which
gave an error of 10.2%.
6-21G was the lowest level of theory used for the geometry optimization.
6-31G was the next highest level of theory used for
the geometry optimization. This proved to be the best level of
theory for geometry optimization as the value came closest to the the
literature3 value of 1.0619 angstroms.
DZV was the highest level of theory used to determine geometry optimization.
This is the highest occupied molecular orbital at
orbital 8. The orbitals were calculated by summing the amount of
electrons in the molecule and dividing by two. In this case the
result was a half-integer so the value was rounded up.
This is the lowest unoccupied molecular orbital at
orbital 9. This would become occupied if the molecule was excited
with the proper amount of energy.
This is the electrostatic potential of the
molecule. The red area represents the lowest electrostatic
potential and blue represents the highest electrostatic potential.
Intermediate colors represent intermediate potentials.
The partial atomic charge on each atom is shown in
this diagram. They are created by the asymmetric distribution of
electrons in a chemical bond.
Table 1 shows the different
orbitals for the NO molecule starting with the S bonding orbitals and
going down to the highest energy orbital. The two P orbitals
represent the px and py orbitals that contribute to pi bonding.
Table 1: Orbitals corresponding to the type of bonding occurring at that level.
| Type of Bonding |
Orbital |
| S sigma bonding |  |
| S sigma anti-bonding | 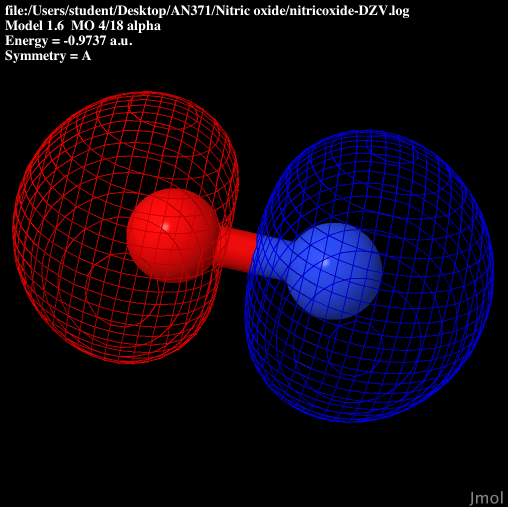 |
| P bonding |
 |
| P anti-bonding | 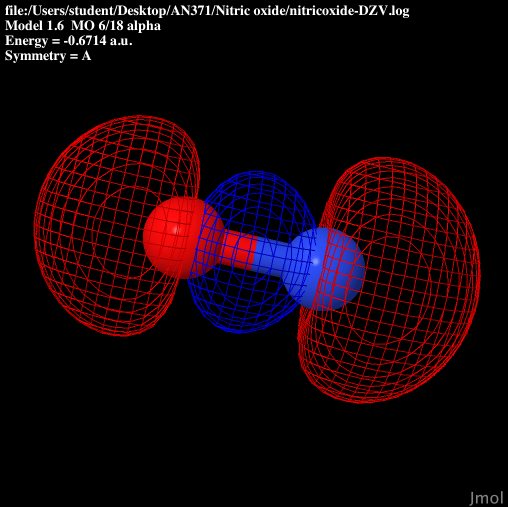 |
| P2 bonding |
 |
| P2 anti-bonding |
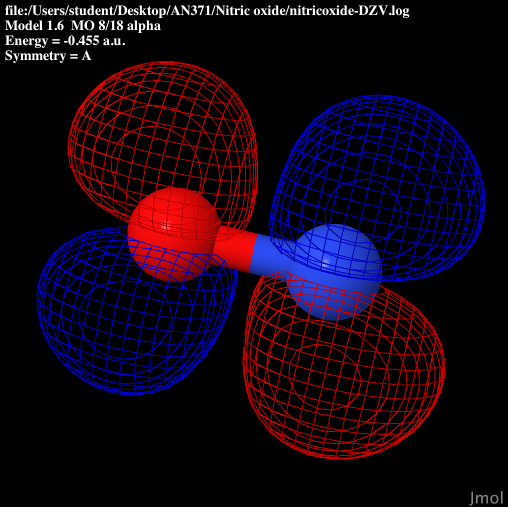 |
The following graph, figure 1, shows the different potential energies of bond stretching at
different levels of theory. The higher the level of theory the lower in
energy the theory calculates for the lowest potential energy. The "bump" as
the potential energy comes out of the well is due to interactions
between the electrons that are not accounted for in the theories. The
experimental graph for the potential energy would not have these "bumps."
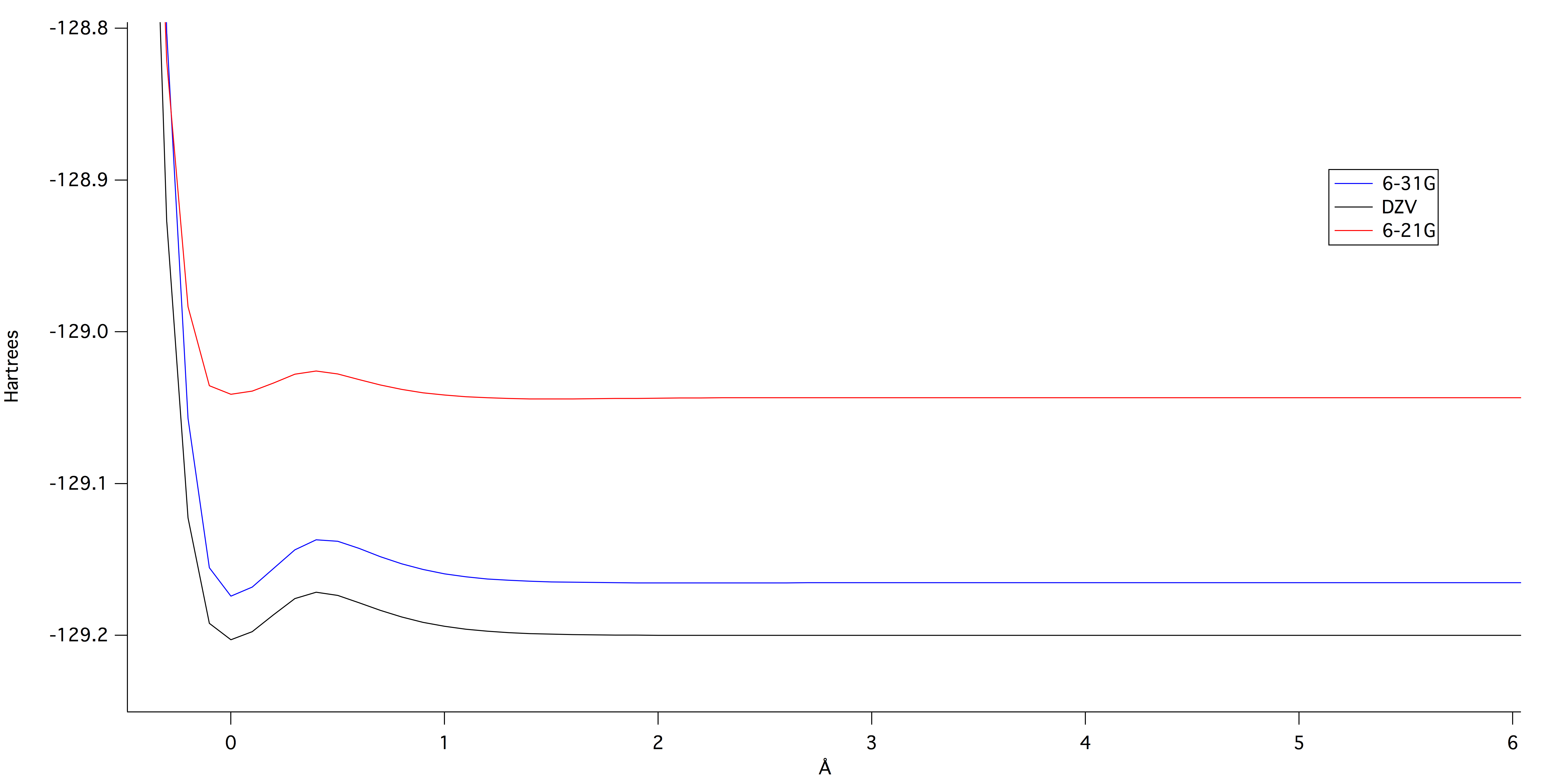
Figure 1: Potential energy curves at
different levels of theory. Note the "bumps" as the curve comes
out of the well. This graph was generated by IGOR.
The final calculated value for
the vibrational frequency using DZV theory came out to be 1158.13
cm^-1. The NIST website3 for nitric oxide gave a value of 1904
cm^-1, which is a percentage error of 39%.
The molecules were calculated at the different levels of theory to determine their dipole moments. They were then compared to the literature value found on NIST website of .153 debyes. The calculated results are shown in table 2.
Table 2: Calculated
dipole moments using different levels of theory. The percent error
was based off the literature value of .153 debyes.
| Theory |
Dipole Moment in Debyes (Db) |
Percent error (%) |
| AM1 |
.090731 |
40.7 |
| 6-21G |
.321852 |
110.4 |
| 6-31G |
.131573 |
14.0 |
| DZV |
.136853 |
10.6 |
You may look at any of these intermediate views again by clicking on
the appropriate button.
Page skeleton and JavaScript generated by export to web function using Jmol 14.1.8 2014-02-10 21:43: on Mar 4, 2014.
This will be the viewer
If your browser/OS combination is Java capable you will get snappier performance if you
use Java.