
Diatomic Oxygen
AM1 Basis Set: When applying the basis
sets to diatomic oxygen, two different electron
configurations were available; singlet and triplet. The
triplet state always appeared to yield the lowest energy
geometries of the diatom, and was continually used and
referenced throughout the course of later calculations.
Molecular geometry optimizations were calculated for diatomic oxygen using several different basis sets. The first and most basic basis set was AM1, a MOPAC2 semi-empirical method. This optimization can be visualized using the button below.
Molecular geometry optimizations were calculated for diatomic oxygen using several different basis sets. The first and most basic basis set was AM1, a MOPAC2 semi-empirical method. This optimization can be visualized using the button below.
|
|
The next three molecular geometry optimizations were calculated on diatomic oxygen using ab initio methods.
6-21G Basis Set:
|
|
6-31 Basis Set:
|
|
Double Zeta Valence Basis Set:
|
|
The DZV set, as seen above, gave a bond length of 1.21 Å. This value was the closest approximation to the experimental bond length found, which was 1.208 Å.1
There are two HOMO orbitals in triplet diatomic oxygen both identical, yet orthogonal to each other with respect to the linearity of the molecule. The two buttons below iterate the orthogonality of the two HOMO states with respect to each other. Observing the animation shows that this orbital is a bonding orbital as the occupied region is shared between the nuclei.
HOMO Orbital 1:
|
|
HOMO Orbital 2:
|
|
The LUMO state of the diatom is an antibonding orbital.
LUMO Orbital:
|
|
There was only one available mode of vibration for the diatomic oxygen molecule.
Vibrational Frequency:
|
|
The valence energy levels of diatomic oxygen were calculated using the DZV basis set. Diagrams of these energy levels can be seen below in Table 1.
| Table 1:
Valence Energy Level Diagram for Diatomic Oxygen |
||
| Energy (a.u.) |
Bond Type |
Molecular Orbital Diagram (from DZV) |
| -20.7604 |
Bonding |
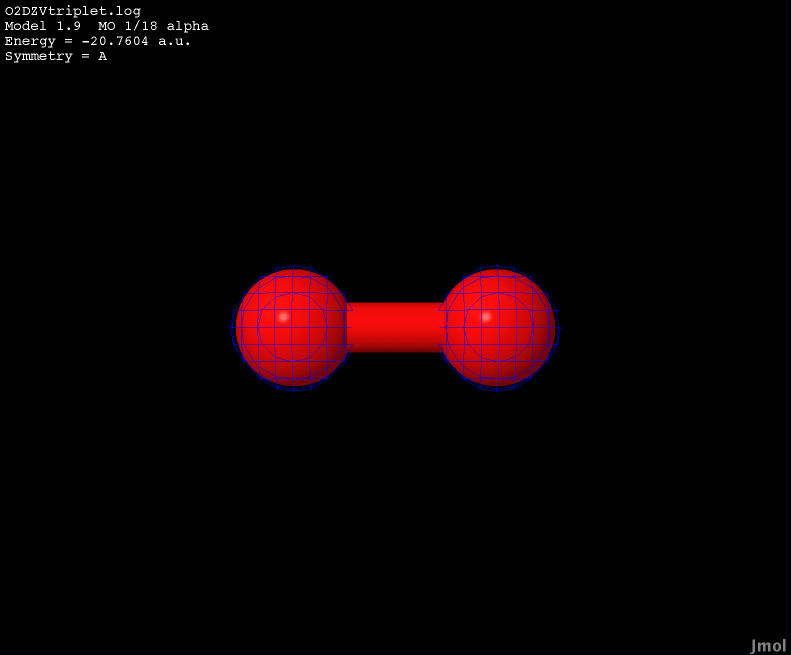 |
| -20.7598 |
Anti-bonding |
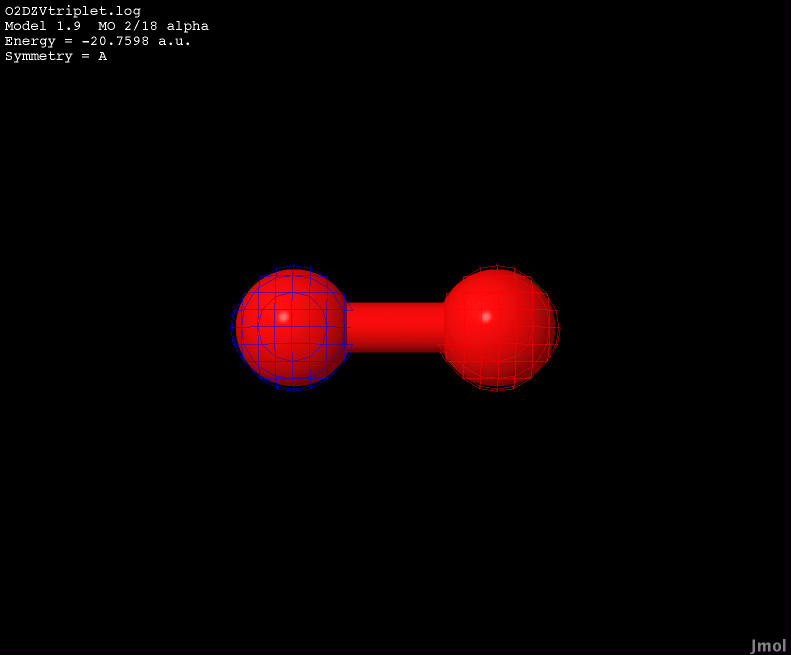 |
| -1.761 |
Bonding |
 |
| -1.2029 |
Anti-Bonding |
 |
| -0.857 |
Bonding |
 |
| -0.857 |
Bonding |
 |
| -0.7544 |
Bonding |
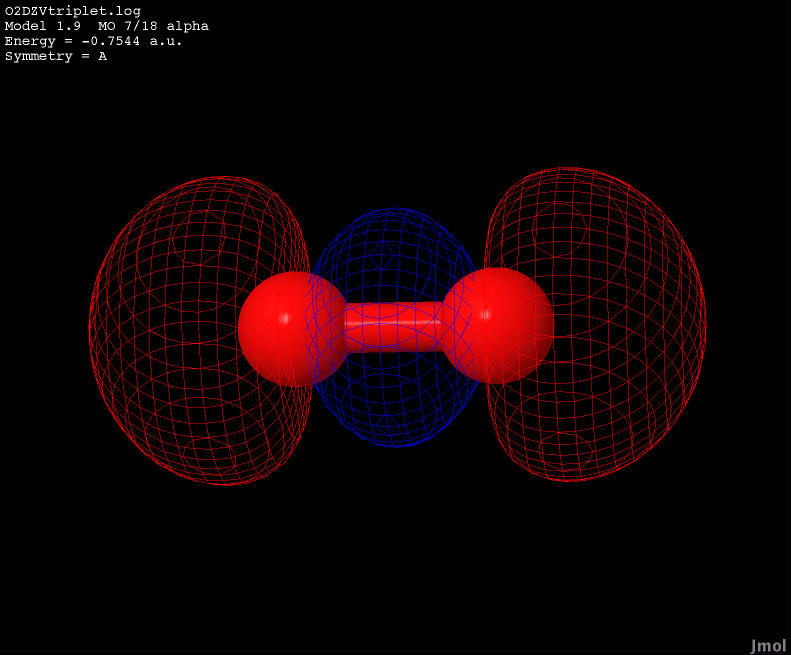 |
| -0.5715 |
Anti-bonding |
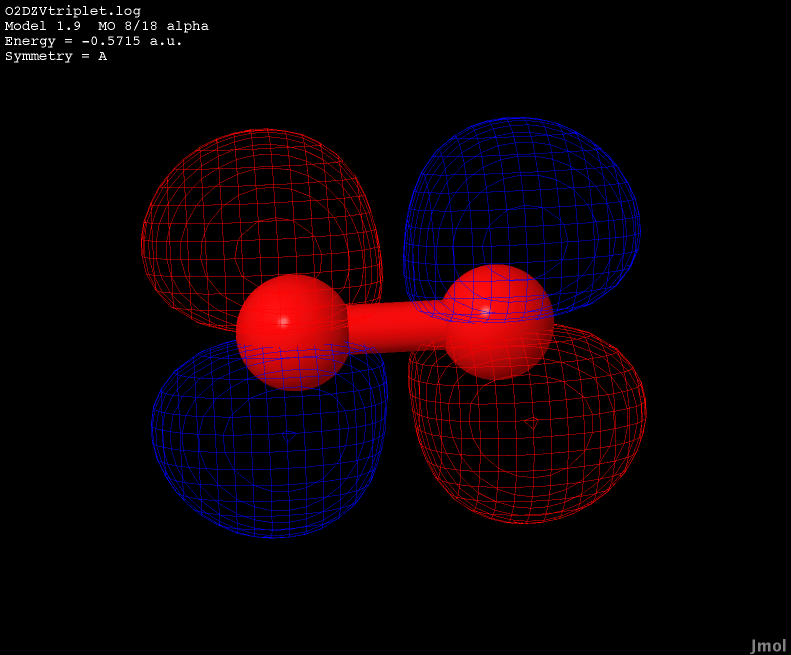 |
| -0.5715 |
Anti-bonding |
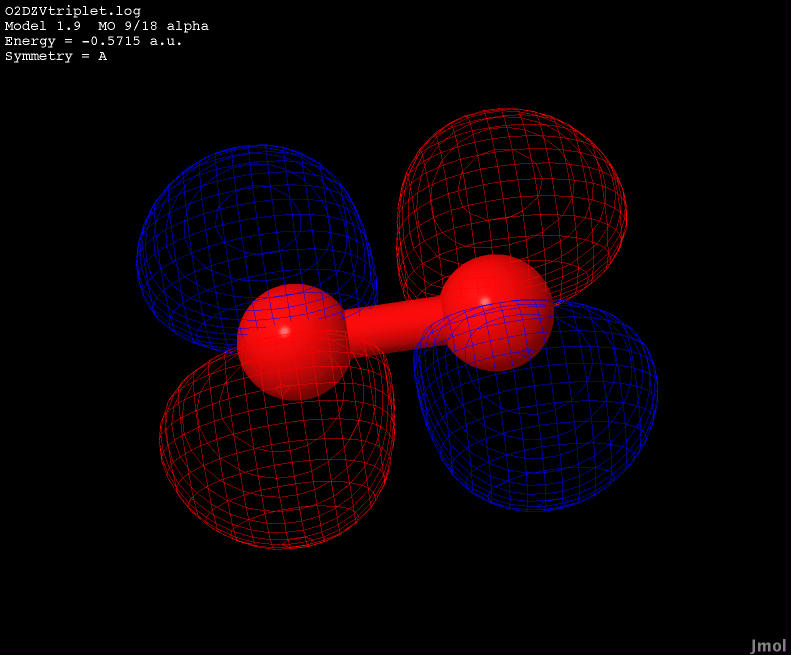 |
| 0.3561 |
Anti-bonding |
 |
| 0.7458 |
Bonding |
 |
| 0.7458 |
Bonding |
 |
| 0.7935 |
Anti-bonding |
 |
| 0.8415 |
Anti-bonding |
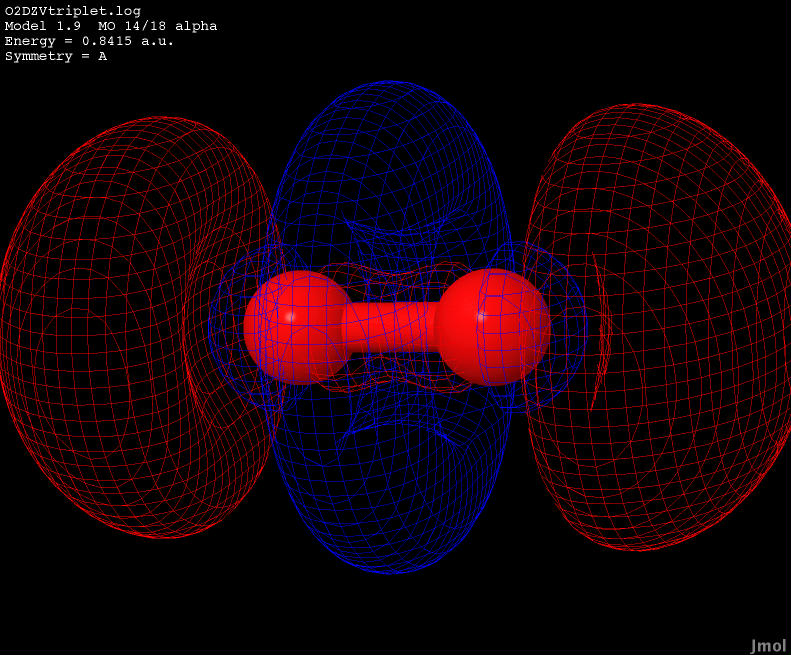 |
| 0.8512 |
Anti-bonding |
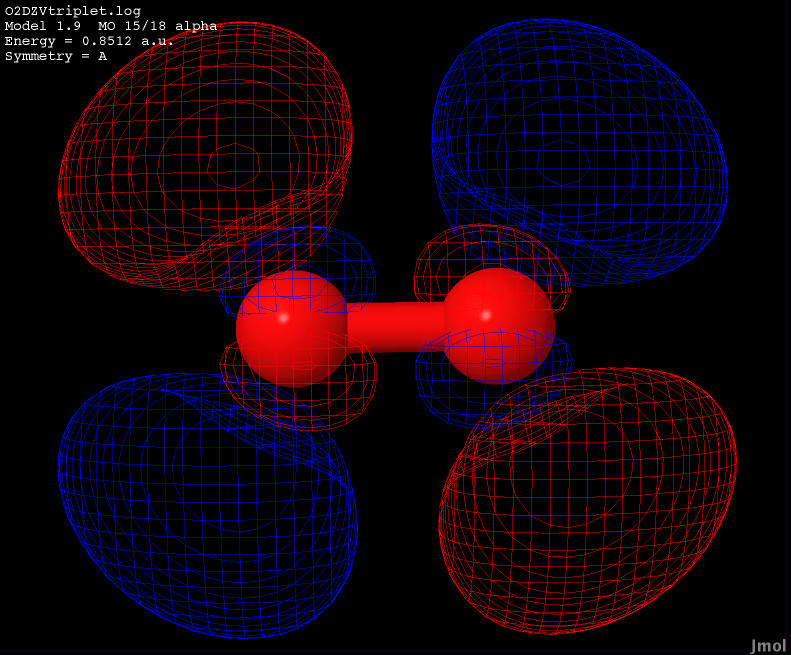 |
| 0.8512 |
Anti-bonding |
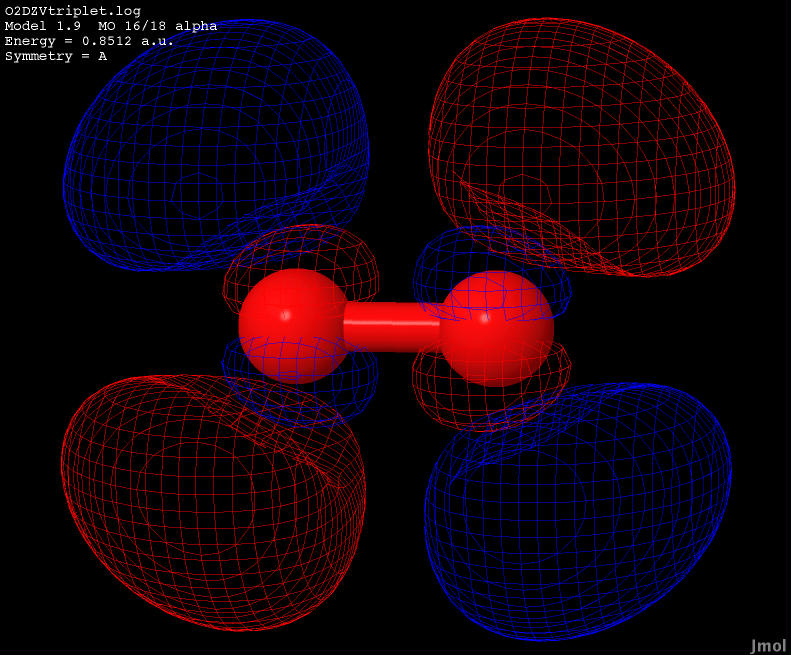 |
| 1.2101 |
Bonding |
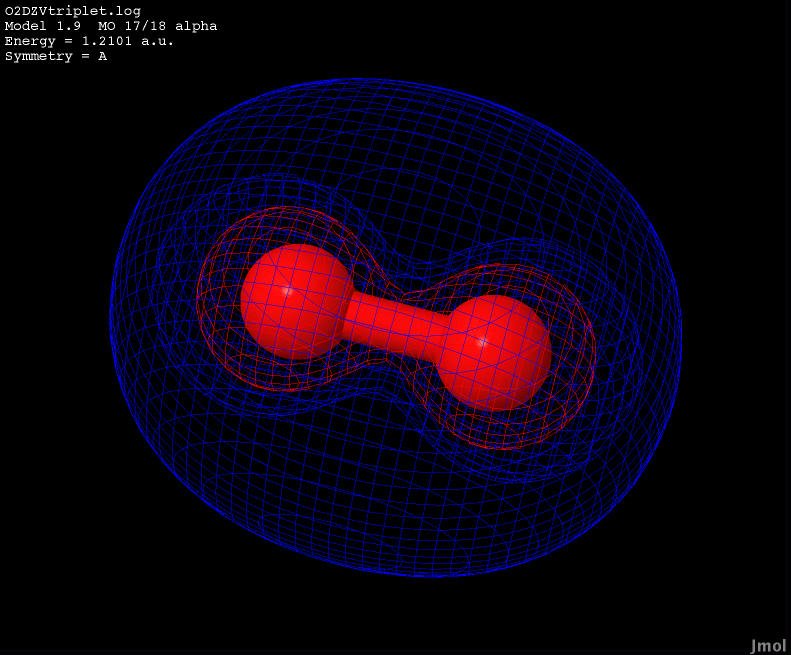 |
| 1.8882 |
Anti-bonding |
 |
A plot of the potential energy of bond stretching for the three ab initio calculations can be seen below in Figure 1. The three basis sets gave nearly identical values for the potential energy, but the 6-21G basis set had the lowest energy initially.
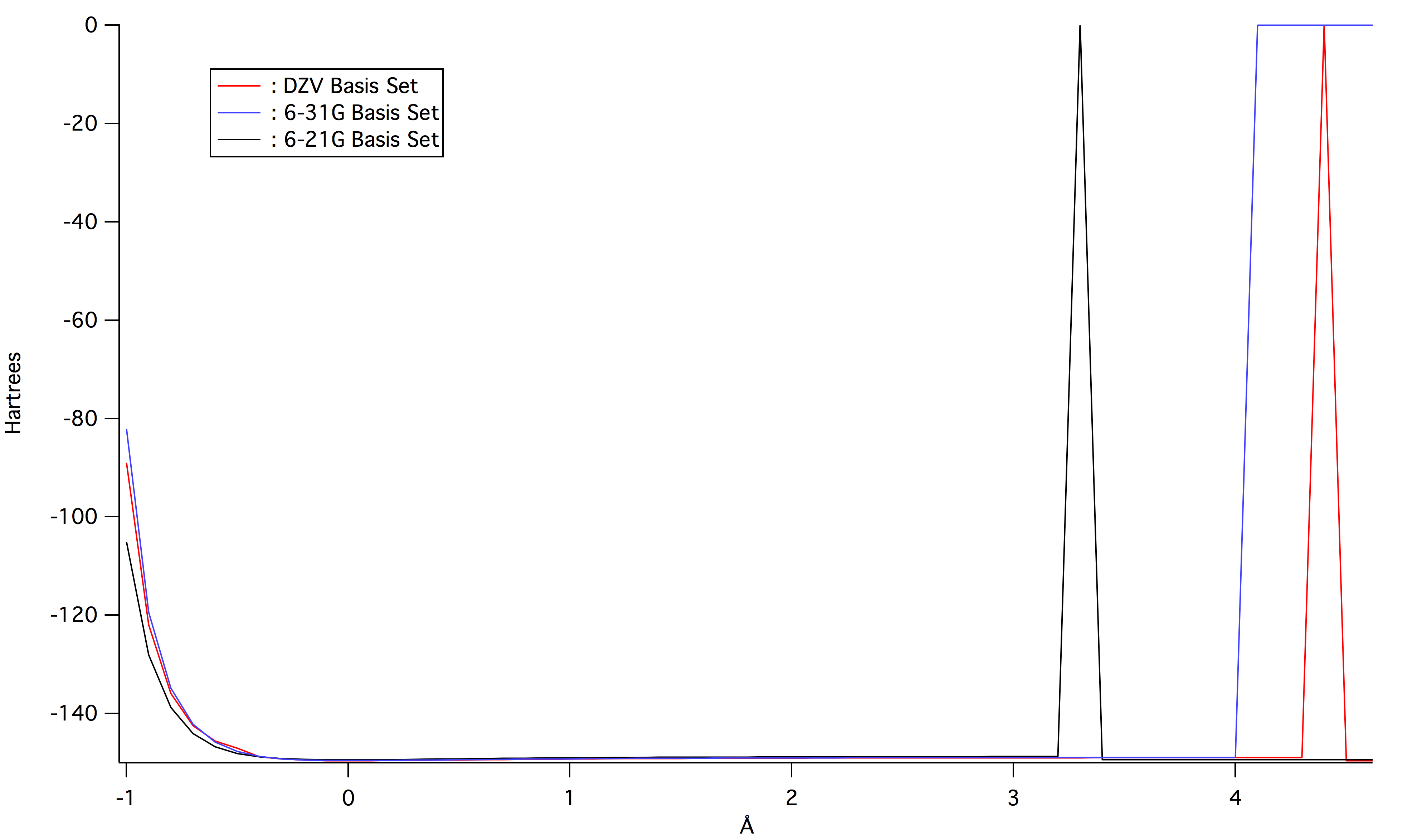
Figure 1: Potential energy of bond stretching of diatomic oxygen for the three ab initio calculations
References
1. Lide D. R., Kehiaian, H.V. CRC Handbook of Chemistry and Physics Data,
CRC Press, Boca Raton, FL, 1992, p F-217.
Page
skeleton and JavaScript generated by export to web function
using Jmol 12.2.34
2012-08-09 20:37 on Feb 26, 2013.