Authors: Casey Freiherr and Anthony Greco
Abstract
Introduction
Many of the physical properties of a single molecule can be
determined by just examining its structure.
The use of the variational principle will allow us to produce a true
wave function with a set of coefficients that give the lowest energy for the
arrangement of electrons, thus providing the most accurate model to obtain
physical data from the molecule.
![]() (1)
(1)
However, the trial wavefunction is not an eigenfucntion of
the Hamiltonian, so the expectation value of energy was necessary to be
calculated.
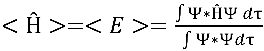 (2)
(2)
Different and larger basis were used to enhance the accuracy
of the predicted energy calculations. To
provide further minimization of energy of the molecules, geometry optimization calculations
were performed with the use of the program Avogadro.
Experimental
A Mac computer was used to construct the following structures using the Avogadro program: carbon monoxide, carbon disulfide and benzaldehyde. Molecular mechanics optimization was performed on each structure using the MMF94s force field. If the molecule was not approaching the right geometry, the molecule could be manually manipulated. Once stabilized, the structure was saved as a .xyz file. WxMacMolPlt was used to generate AM1 and PM3 geometry optimization input files (.inp files) for the GAMESS computation package. GamessQ was used to submit these into GAMESS. After each optimization, the logs were viewed to make certain that the molecules had exited gracefully. If they had not, the structure had to be checked to correct any errors. If they did, the file was saved in each molecule's directory for future access. Each of the working result files (.log), the geometry optimization was check to ensure it was completed. Once optimized, each molecule was optimized further using higher levels of theory starting at 621-G, then going to 631-G and finally DZV, or double zeta valence. The lowest level was run first with the next level of theory using the previous as the basis. Each computation was completed, they were checked to ensure they were successful. Using the final successful level of theory, properties of the molecules could be calculated. Properties of interest were the bond lengths, vibrational frequencies, bond angles, dipole moments, partial atomic charges UV-Vis transition energy and the potential energy surface. The dipole moments were able to be found using the logs of the optimized geometry and searching for "Debye". Bond lengths and angles were able to be found using the optimized geometry in Jmol and selecting the specific atoms of interest. Jmol also had all the possible orbitals the complex could offer. The HOMO was calculated by summing the number of electrons each atom had and dividing by two. The LUMO is then above the HOMO. For the vibrational frequencies, the accepted IR spectrum for each structure was used to find the specific peaks. Then, using Jmol, the structures at those specific frequencies were taken and reported in the links below.
| Carbon Monoxide |
Carbon Disulfide |
Benzaldehyde |
 |
 |
 |
| Click on image to go to the CO page. |
Click on image to go to the CS2 page. |
Click on image to go to the benzaldehyde page. |
Conclusions
After using the results from each of the molecules, there have been discrepancies from the data that was calculated to the data resulting from experiments. This procedure can be useful as a starting point for an experiment as an estimate for certain values such as bond length or geometries. This can also be useful as a prediction for where peaks can be found in IR or UV-Vis spectra. However, this should not be completely relied upon for a trusted value for energies due to the Hartree-Fock Self-Consistent Field calculations not coupling electrons resulting in error due to the electrons being forced together closer than what they actually are on average. All experimental values should take priority before these calculations be published as an accepted value.
References
(1) Mihalick, J.; Gutow, J. Molecular Orbital Calculations. Oshkosh, WI, 2014.
Cooksy, Andrew. "Hartree-Fock SCF." Physical Chemistry. Quantum Chemistry and Molecular Interactions. pag. 174-178 Print.