
The button for optimized geometry appears below. The JMOL calculations feature was used to find the optimized geometry for Br2
The button below shows the optimized geometry for the Br2 molecule. The bond length was calculated with JMOL.
The button below will show the lowest unoccupied molecular orbital (LUMO). The LUMO was found using the log files compiled using GAMESSQ.
The button for the highest occupied molecular orbital (HOMO) is shown below. The HOMO was found using the log files computed by GAMESSQ.
The button for the electrostatic potential of this molecule was found using the DZV surface log files computed by GAMESSQ. JMOL was used to draw the electrostatic potential.
The button for partial atomic charges is shown below. They were calculated with GAMESSQ.
The dipole moment was calculated to be 0 for all levels of theories. This makes sense since it is a non-polar molecule. The molecule consists of only 2 Br molecules, which have the same electronegativity.
This table shows the valence electron configurations for the molecular orbitals in increasing order of energy.
| Valence Energy Level | Molecular Orbital |
| 1s sigma bonding |  |
| 1s sigma anti bonding |  |
| 2s sigma bonding | 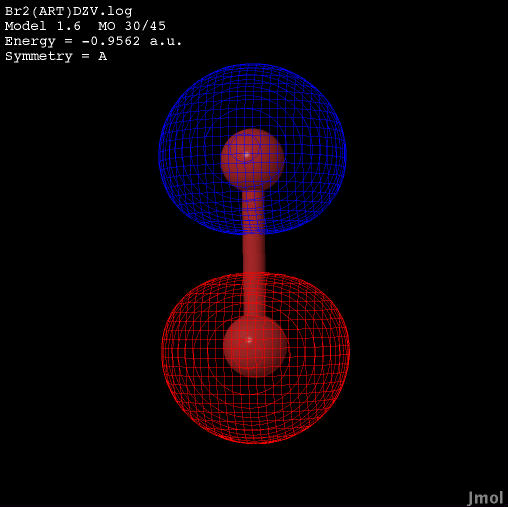 |
| 1 pi bonding |  |
| 1 pi anti bonding |  |
The potential energy vs bond length is shown below for the 6-21G energy level. This graph was created using the log file for the 6-21G energy level and a macro in IGOR. We weren't able to make the other energy levels like 6-31G and DZV work with the IGOR macro.
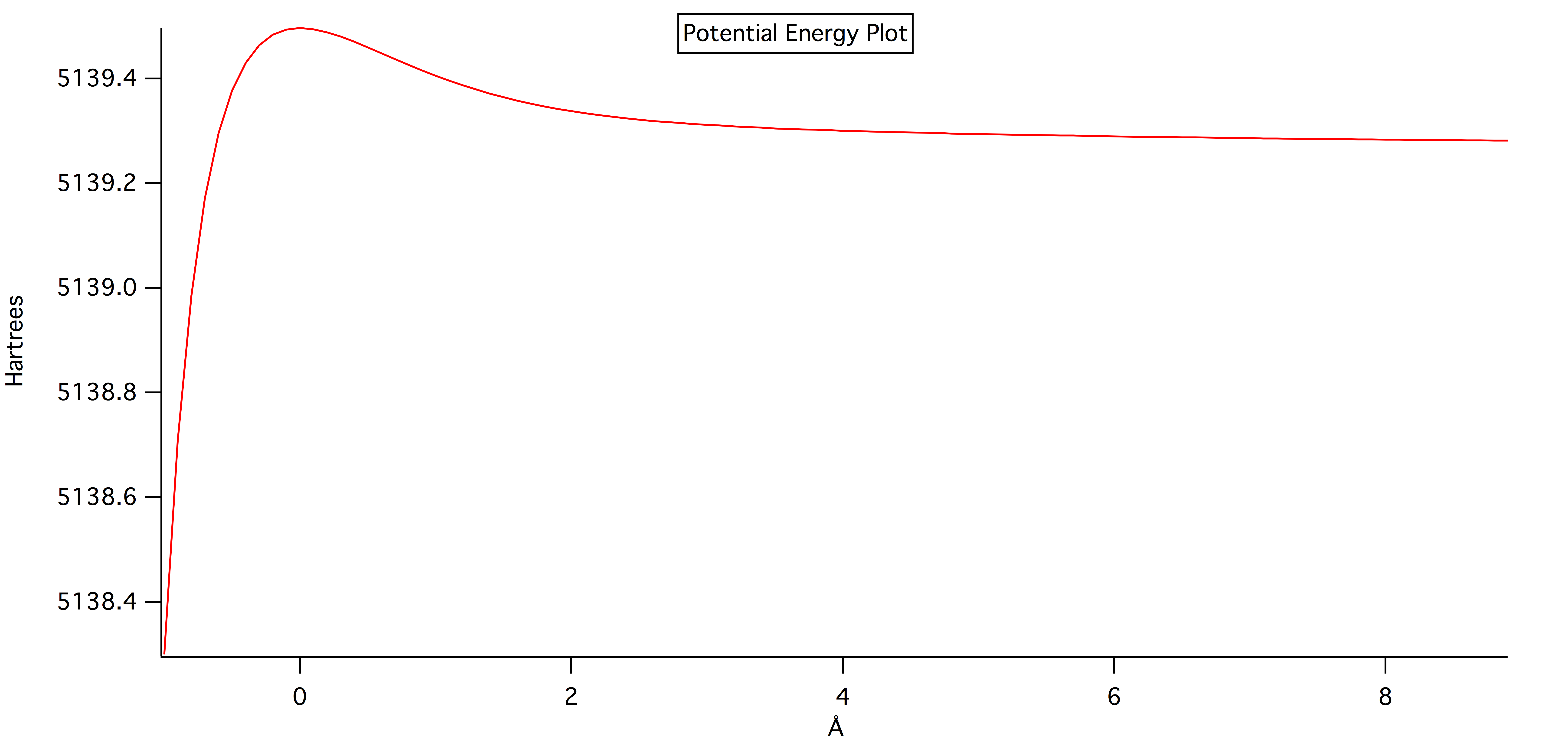
The calculated vibrational frequency was found to be 310.01cm-1 using JMOL and the vibrational calculations calculated with GAMESSQ.
Page skeleton and JavaScript generated by export to web function using Jmol 12.2.16 2011-12-13 21:20 on Mar 4, 2012.