Potential Energy of Bond Stretching
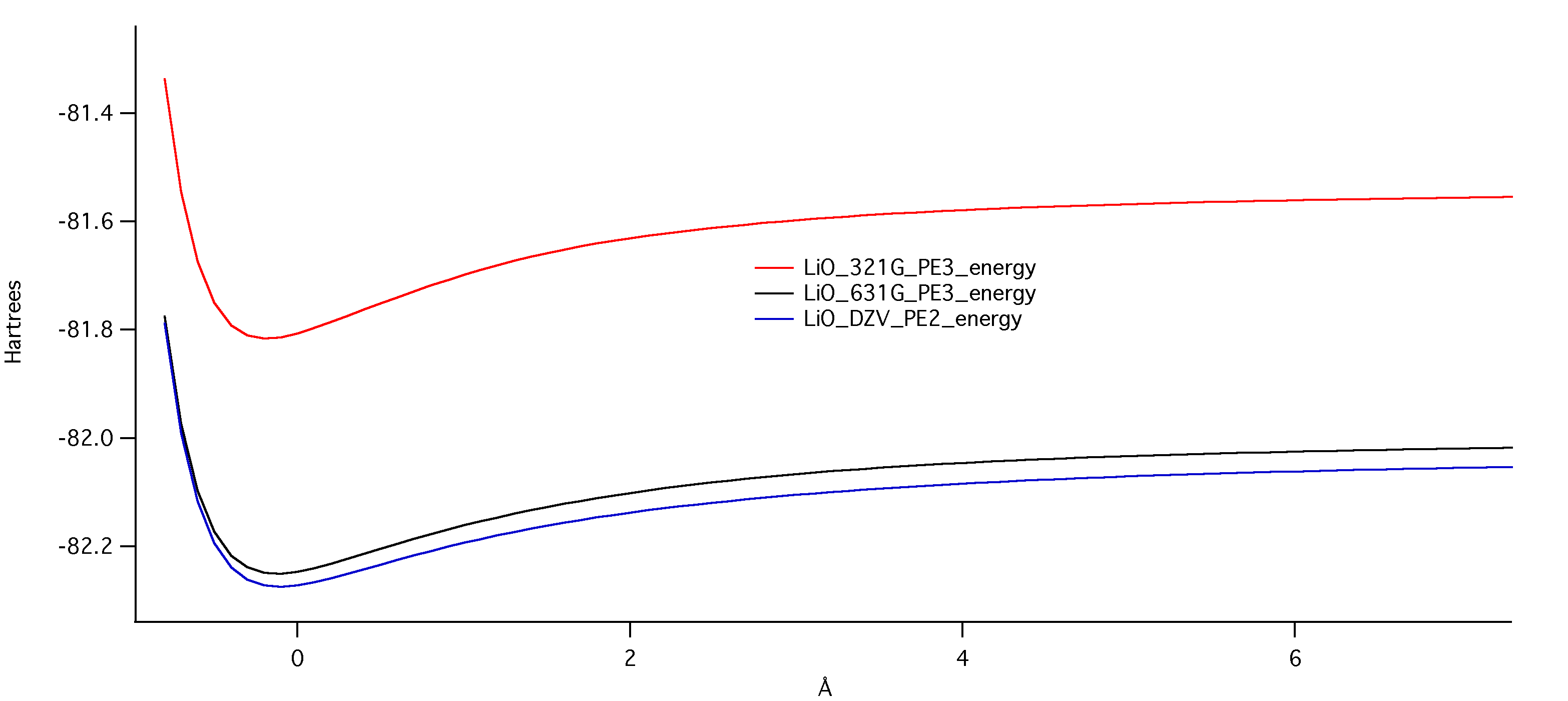
The graph below shows the potential energy for all three levels of theory. The DZV level (the blue line) has the lowest potential energy, and thus is the best approximation for opitmized geometry. This is yet another indication that bigger basis sizes are needed for best results.
Vibrational Frequency
The vibrational frequency data calculated only included one vibrational peak, at 816.23cm^-1.
Getting a Better Dipole
Dipole moments were obtained from a previous experiment, but then were recalculated with diffuse functions in order to get a better dipole. The calculation was run three times, each with a different number of polarization functions for D heavy atoms and light atoms (represented by 111, 212, and 313). The number for F heavy atoms, which is the central number, was required to be always 1. The electrostatic moments and partial charges are shown below for each calculation.
Electrostatic Moments: X, Y, Z in bohr; DX, DY, DZ in debye.
| X |
Y |
Z |
Charge(AU) |
DX |
DY |
DZ |
/D/ |
|
| 111 |
0 |
0 |
.066962 |
.00 |
0 |
0 |
-6.923281 |
6.923281 |
| 212 |
0 |
0 |
.066962 |
.00 |
0 |
0 |
-6.905664 |
6.905664 |
| 313 |
0 |
0 |
.066962 |
.00 |
0 |
0 |
-6.992058 |
6.992058 |
Partial Charges
111
| Atom |
Mull. Pop. |
Charge |
Low. Pop. |
Charge |
| Li |
2.335124 |
.664876 |
2.778553 |
.221447 |
| O |
8.664876 |
-.664876 |
8.221447 |
-.221447 |
| Atom |
Mull. Pop. |
Charge |
Low. Pop. |
Charge |
| Li |
2.370560 |
.629440 |
2.814017 |
.185983 |
| O |
8.629440 |
-.629440 |
8.185983 |
-.185983 |
| Atom |
Mull. Pop. |
Charge |
Low. Pop. |
Charge |
| Li |
2.399572 |
.600428 |
2.810366 |
.189634 |
| O |
8.600428 |
-.600428 |
8.189634 |
-.189634 |
The results here are not necessarily better with bigger basis sets. It turns out that the number of functions does not matter so much with dipole moments -- better results are obtained through the addition of a few large and specific polarization functions.
Literature Comparison: The literature3 gave an experimental value of 6.843 in the Debye. The calculated dipole moment is greater than this, but only by about 1%.