
By: Peter and Alina
Molecular orbital calculations have historically been a rather involved process requiring an intimate knowledge of quantum mechanics. Modern computation methods allow for chemists and students alike, to do such calculations with a somewhat limited understanding of quantum properties. These quantum calculations were done with various levels of theory using MacMolPlot and GTK GAMESS software programs. On these software packages optimal geometry, vibrational, and HOMO calculations were performed on diatomic chlorine, ozone (O3) and Benzoic acid.
The best results for chlorine were obtained using Double Zeta optimization as the highest level of theory. This conclusion was reached by observing the potential energy curves at three levels of theory including 3-21G, 6-31, and Double Zeta Valence. This comparison shown below gives the Double Zeta Valence calculation having the lowest energy bond length.
The optimized bond length for Cl2 was calculated to be 1.99 Angstroms compared to the literature (NIST) value of 1.988 Angstroms.
The Highest Occupied Molecular Orbital (HOMO) for chlorine was calculated and the probability density is shown here.
The vibration of the bond of Cl2 moves the molecule in a inward pulsing fashion. The frequency given by this vibration is 668.62 cm-1 which is rather close to its literature value of 559.7 cm-1
Ozone
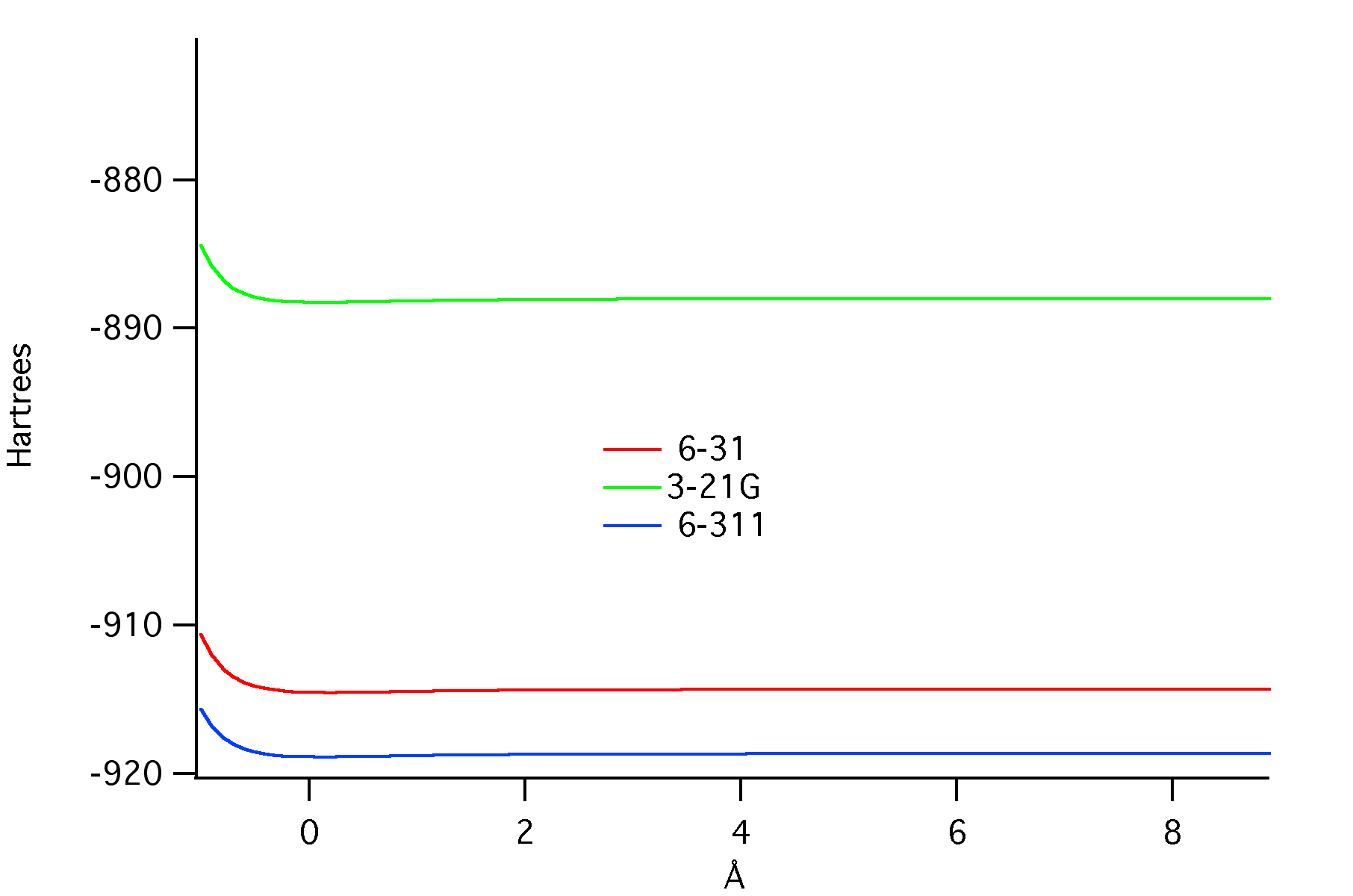
Calculations were also performed on Ozone (O3) with three levels of theory including 3-21G, 6-31, and 6-311. With the 6-311 theory level being the highest and thus the most optimal geometric results.
Each bond length of O3 was calculated to be 1.16 Angstroms and can be accurately compared to the literature value of 1.218 Angstroms.
120.9 degrees is the measurment of the bond between the Oxygen atoms in O3. The literature value obtained was 116.8 degrees which is rather similiar.
The first vibration looked at with O3 is a symmetric vibration where the oxygen atoms at the end of the molecule are moving outward away from the middle atom. The frequency of this is 1333.74 cm-1, compared to a literature value of 1110 cm-1.
The Asymmetric vibration for O3 moves in the fashion of the middle oxygen atom pushing up from left to right toward the outer atoms. The literary value is 1042.1 cm-1 which is extremely close to the calculated value of 1019.1 cm-1. Spectral data also obtained from NIST shows a definite peak at 1020 cm-1.
The frequency of the scissoring vibration of O3 is 742.02 cm-1 which also comes very close to the literary value of 705 cm-1. The movement of this vibration is pretty much self explanitory, it moves the same way scissors do.
Reference Specra can be viewed on the online NIST database.
The Highest Occupied Molecular Orbital (HOMO) for ozone was calculated at all levels of theory, however the 6-311 with optimal geometry is shown here
The energy for the HOMO of O3 is -0.0515.
Benzoic Acid
The same levels of theory used in the calculations for ozone (3-21G, 6-31, 6-311) were performed on benzoic acid again having the 6-311 level of theory being the highest. From this calculation the optimal bond lengths, bond angles and UV absorbance data were obtained.
There are many different bond lengths associated with Benzoic Acid ranging from 0.97 to 1.4 Angstroms.
Bond angles also vary throughout Benzoic Acid depending on the type of bond. These angles range from 109.2 to 120.8 degrees.
The carbonyl vibration was calculated to be 1877.97 cm-1 which closely resembels the spectral data with a sharp peak around 1750 to 1775 cm-1.
The OH vibration was calculated to be 4077.54 cm-1 which is simmilar to the spectral data which shows a peak at 3575 cm-1.
The Phenyl vibrational mode shown here is spectrally located in the fingerprint region.
The Highest Occupied Molecular Orbital (HOMO) for benzoic acid was calculated at all three levels of theory but the the 6-311 level gve optimal results.
The energy for the Benzoic Acid HOMO was calculated to be -0.3539
UV Transitions for Benzoic Acid
(From ground State)
| 3-21G | (eV) | 6-31 | (eV) | 6-311 | (eV) |
| 2 | 6.28 | 2 | 6.02 | 2 | 5.98 |
| 4 | 8.25 | 4 | 7.99 | 4 | 7.64 |
| 6 | 9.08 | 6 | 8.77 | 6 | 8.48 |
| 8 | 9.37 | 8 | 9.18 | 8 | 8.92 |
Shown above is a table of the transition energies in electron volts, from the ground state to some excited state (2,4,6 and 8) for all levels of theory. The energy required to reach higher levels decreases signifigantly than that required to transition to the first excited state. What can also be observed is that the calculated energy decreases as higher levels of theory are used thus confirming 6-311 as the best optimization.
Reference data can be compared to the NIST online database.
Conclusions
There are plenty of different calculations that can be found in quantum mechanics. A few that were run but not included were potential energy and UV visible spectrum. The software of MacMolPlot and GTK GAMESS were very useful and accurate in finding different features of our molecules. Much of the data found was relatively close to the literature values obtained from NIST.

References:
NIST website
<http://webbook.nist.gov/chemistry>
Page
skeleton and JavaScript generated by export to web function using Jmol 11.6.6 2008-09-20
22:06 on Mar 30, 2009.