
Carbon Monoxide Quantum Calculations
All of the displays on this page were created using our best optimized geometry calculations. The level of ab initio theory used was 6-311G. During the calculation, the geometry was approximated with a "balls on springs" model. Once the calculation was completed, the geometry with the lowest energy conformation was chosen as the optimum.
By clicking on the Bond Length button below, a live display of the optimized geometry for CO will appear with the bond length in nanometers.
The bond length is more precisely 0.112368 nm, which is very close to the experimental value of 0.112830 nm from the NIST website.3 The error is only 0.4%.
Below is a molecular potential energy curve for CO displaying the potential energy in Hartrees and the bond length in Angstroms. The calculations were done using the three different basis set sizes shown in the legend. They are 3-21G, 6-31G, and 6-311G in order of increasing size.
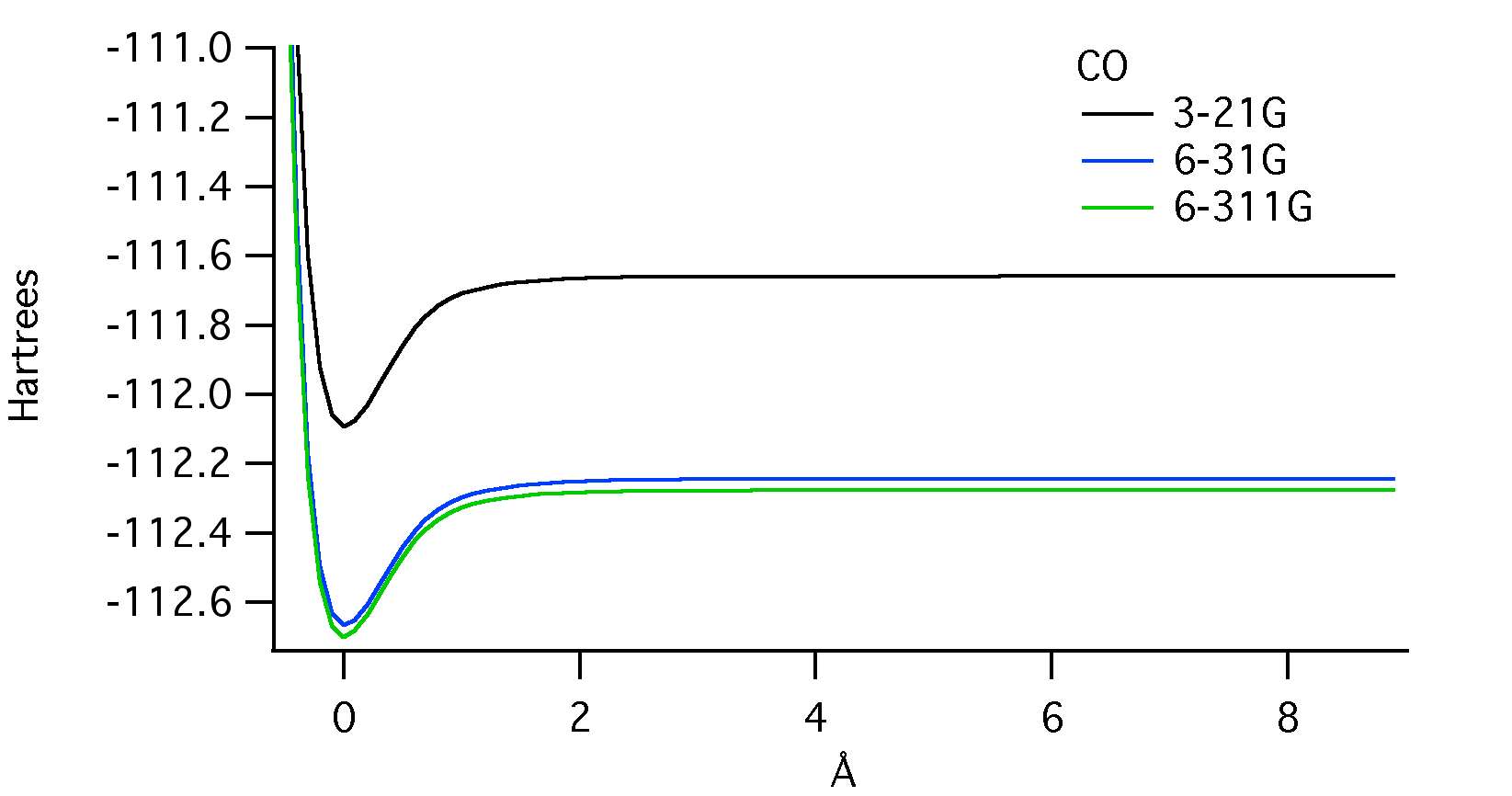
The energy minimum indicates the equilibrium bond length for carbon monoxide.2 The difference in this value between basis set sizes appears to be negligible. The energy at which the internuclear separation occurs (where the curves level off), however, is obviously different between basis set sizes. The energy for the 6-31G calculation is about 0.05 Hartrees more than the 6-311G, and the 3-21G calculation is about 0.6 Hartrees more. The depth of the energy minimum is closely related to the bond dissociation energy.2 This value, just like the bond length, appears to be very similar between basis set sizes.
The Vibration button below will display carbon monoxide's single mode of vibration.
The calculated frequency of this vibration is 2281.09 cm-1. The experimental value from NIST3 is 2169.80 cm-1, which gives an error of about 5%.
For an IR spectrum of CO, visit the NIST webpage.4 It is apparent from the spectrum that our calculation of vibration is too high.
For a display of carbon monoxide's HOMO, click the button below.
According to the program MacMolPlt, the orbitals displayed are mostly S and Px in character from both the carbon and oxygen and contribute to bonding.
Page skeleton and JavaScript generated by export to web function using Jmol 11.6.6 2008-09-20 22:06 on Mar 17, 2009.