Once the molecule file is fully loaded, the image at right will become live. At that time the "activate 3-D" icon
![]()
will disappear.
Ortho-xylene
The best geometry for the highest level of theory, DZV, was used to
calculate the bond lengths and bond angles. The bond length for the C-C
bonds are 1.39-1.41 angstroms, C-H bonds are 1.7-1.8 angstroms, and the
C-Me bonds are 1.52 angstroms. The bond angles were all around 119-120
degrees and the hydrogens on the methyl groups had angles about 107-110
degrees.
The HOMO and LUMO where then calculated and diagrammed below.
The highest occupied molecular orbital for
o-xylene is at 29. This is calculated by the sum of electrons (58)
divided by two.
The lowest unoccupied molecular orbital for
o-xylene is then calculated to be 30, which is one more orbital that the
HOMO.
The partial atomic charges and electrostatic potential are also calculated and shown in the diagrams below.
The partial atomic charge shown in this
structure is derived from the partial negative and partial positive
charges distributed from the electrons in the bond.
This diagram shows the electrostatic
potential. The blue is the highest potential and the red is the lowest
potential. The green is the intermediate potential.
The vibrational frequencies were calculated from the
highest level theory and best geometry, DZV. The associated IR spectrum
is shown in Figure 1.
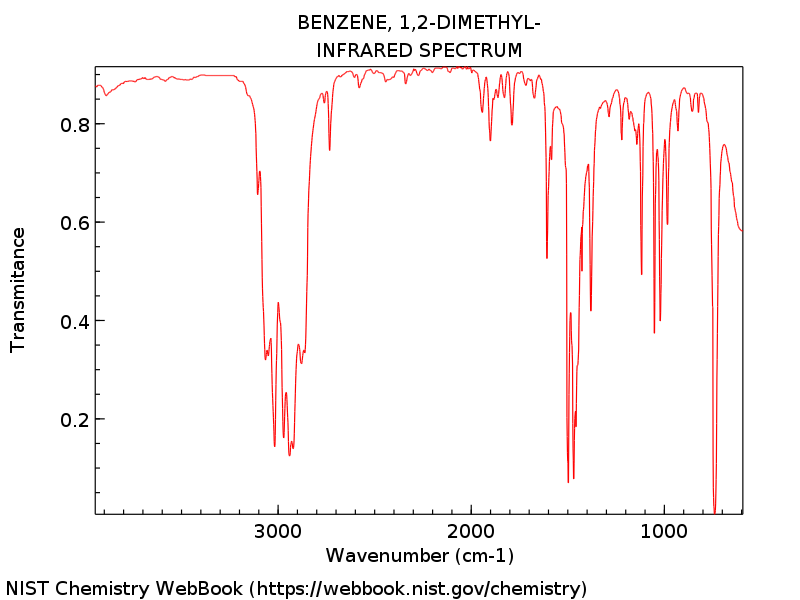
Figure 1: IR spectrum of o-xylene, collected from an outside source1 that ranges from 500-4000 cm-1.
This vibrational frequency is associated with C-Me stretching.
This vibrational frequency is associated with a C-H wag.
This vibrational frequency is associated with a C-Me stretching.
This vibrational frequency is associated with a oscillation of the C-H.
This vibrational frequency is associated with stretching of the aromatic ring.
The UV-Vis peaks were then calculated and are listed in Table 1 below.
Table 1: Lists the oscillation strength and wavelength of o-xylene.
Oscillater Strength
|
Wavelength (nm)
|
1.104008
|
150.49
|
1.428811
|
149.58
|
0.018935
|
127.44
|
A UV-Vis spectrum was not acquired for this molecule. A literature value of 260 nm was obtained2.
Dipole moments were also calculated and compared to a literature value.
Table 2: List of dipole moments for all levels of theory.
Theory Level
|
Dipole Moment (Debye)
|
AM1
|
0.461782
|
PM3
|
0.457608
|
6-21G
|
0.558818
|
6-31G
|
0.553172
|
DZV
|
0.646282
|
The experimental value3 is 0.62 Debyes, which is very close to the calculated DZV dipole moment listed in Table 2.
You may look at any of these intermediate views again by clicking on the appropriate button.
Based on a template by A. Herráez and J. Gutow
If your browser/OS combination is Java capable, you will get snappier performance if you use Java
Using directory /Users/westpl39/Desktop/P-Chem_Documents/Lindsey:Holleigh Molecules/Website/o-xylene/o-xylene
adding support.js
...jmolApplet0
...adding ortho-xylene.png
copying and unzipping jsmol.zip directory into /Users/westpl39/Desktop/P-Chem_Documents/Lindsey:Holleigh Molecules/Website/o-xylene/o-xylene
...adding ortho-xylene.spt
...jmolApplet1
...adding DZV_Bond_Length.png
copying and unzipping jsmol.zip directory into /Users/westpl39/Desktop/P-Chem_Documents/Lindsey:Holleigh Molecules/Website/o-xylene/o-xylene
...adding DZV_Bond_Length.spt
...jmolApplet2
...adding DZV_Bond_Angle.png
copying and unzipping jsmol.zip directory into /Users/westpl39/Desktop/P-Chem_Documents/Lindsey:Holleigh Molecules/Website/o-xylene/o-xylene
...adding DZV_Bond_Angle.spt
...jmolApplet3
...adding HOMO_MO_29.png
copying and unzipping jsmol.zip directory into /Users/westpl39/Desktop/P-Chem_Documents/Lindsey:Holleigh Molecules/Website/o-xylene/o-xylene
...adding HOMO_MO_29.spt
...jmolApplet4
...adding LUMO_MO_30.png
copying and unzipping jsmol.zip directory into /Users/westpl39/Desktop/P-Chem_Documents/Lindsey:Holleigh Molecules/Website/o-xylene/o-xylene
...adding LUMO_MO_30.spt
...jmolApplet5
...adding Partial_Atomic_Charges.png
copying and unzipping jsmol.zip directory into /Users/westpl39/Desktop/P-Chem_Documents/Lindsey:Holleigh Molecules/Website/o-xylene/o-xylene
...adding Partial_Atomic_Charges.spt
...jmolApplet6
...adding Electrostatic_Potential.png
copying and unzipping jsmol.zip directory into /Users/westpl39/Desktop/P-Chem_Documents/Lindsey:Holleigh Molecules/Website/o-xylene/o-xylene
...adding Electrostatic_Potential.spt
...jmolApplet7
...adding 750_IR_Vib.png
copying and unzipping jsmol.zip directory into /Users/westpl39/Desktop/P-Chem_Documents/Lindsey:Holleigh Molecules/Website/o-xylene/o-xylene
...adding 750_IR_Vib.spt
...jmolApplet8
...adding 1000_IR_Vib.png
copying and unzipping jsmol.zip directory into /Users/westpl39/Desktop/P-Chem_Documents/Lindsey:Holleigh Molecules/Website/o-xylene/o-xylene
...adding 1000_IR_Vib.spt
...jmolApplet9
...adding 1100_IR_Vib.png
copying and unzipping jsmol.zip directory into /Users/westpl39/Desktop/P-Chem_Documents/Lindsey:Holleigh Molecules/Website/o-xylene/o-xylene
...adding 1100_IR_Vib.spt
...jmolApplet10
...adding 1375_IR_Vib.png
copying and unzipping jsmol.zip directory into /Users/westpl39/Desktop/P-Chem_Documents/Lindsey:Holleigh Molecules/Website/o-xylene/o-xylene
...adding 1375_IR_Vib.spt
...jmolApplet11
...adding 1600_IR_Vib.png
copying and unzipping jsmol.zip directory into /Users/westpl39/Desktop/P-Chem_Documents/Lindsey:Holleigh Molecules/Website/o-xylene/o-xylene
...adding 1600_IR_Vib.spt